



前面说了如何用eggNOG-mapper快速注释功能基因,我们最后得到了很多结果文件,其中最重要的是两个annotations文件。这里主要讲一下怎么整理结果文件,并且对注释的结果做质量评估。
1. 基因功能注释评估
其实就是整理结果文件中有多少基因注释到了哪些数据库中,以一种直观的方式展现结果。
这里分两种情况,如果自己喜欢以编程的方式处理文件,可以直接处理out.emapper.annotations这个文件。如果前一种方式不是很得心应手的话,那就直接处理out.emapper.annotations.xlsx这个文件,毕竟可以用excel直接打开。
在上一篇博客中详细介绍了结果文件中20列代表什么含义,删掉前两行运行的参数和命令,剩下的就是我们熟悉的excel表格。
将第一行表头的内容选中,筛选。A列(query)就是eggNOG注释到的基因名,选中并且粘贴到新的表格;J列(GOs)筛选除了“-”以外的内容,复制第一列作为注释到GO的序列;L列(KEGG_ko)筛选除了“-”以外的内容,复制第一列作为注释到KEGG的序列;U列(PFAMs)筛选除“-”以外的内容,复制第一列作为注释到PFAM的序列。可以得到如下形式的表格(另存为annotation.xlsx):
这种类型的输入数据可以在本地导入R包做韦恩(Venn)图,也可以选择用在线工具直接生成韦恩图,比如这个网站:Venny 2.1.0 (csic.es)
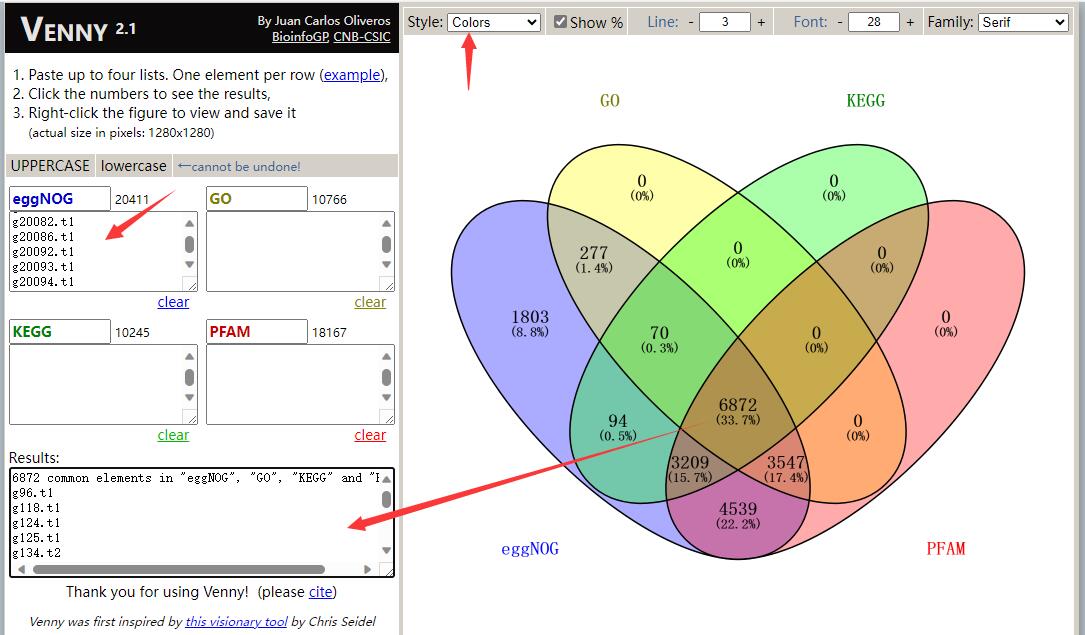
最多支持四组样本,把数据贴进左边4个框内即可。上方的Style可以选择不同的着色方式,点击图中的数字可以在左下方Results框内看到各个数据集的重叠关系,右键图片也可以直接保存。这个功能不难实现,有空可以更新到我的本地小工具集合中。
类似的在线做韦恩图的网站还有很多:
或者选择自己折腾R,用经典的VennDiagram包做韦恩图,或者用UpSetR包做Upset图:
1 | # R包readxl导入xlsx文件,VennDiagram做Veen图 |
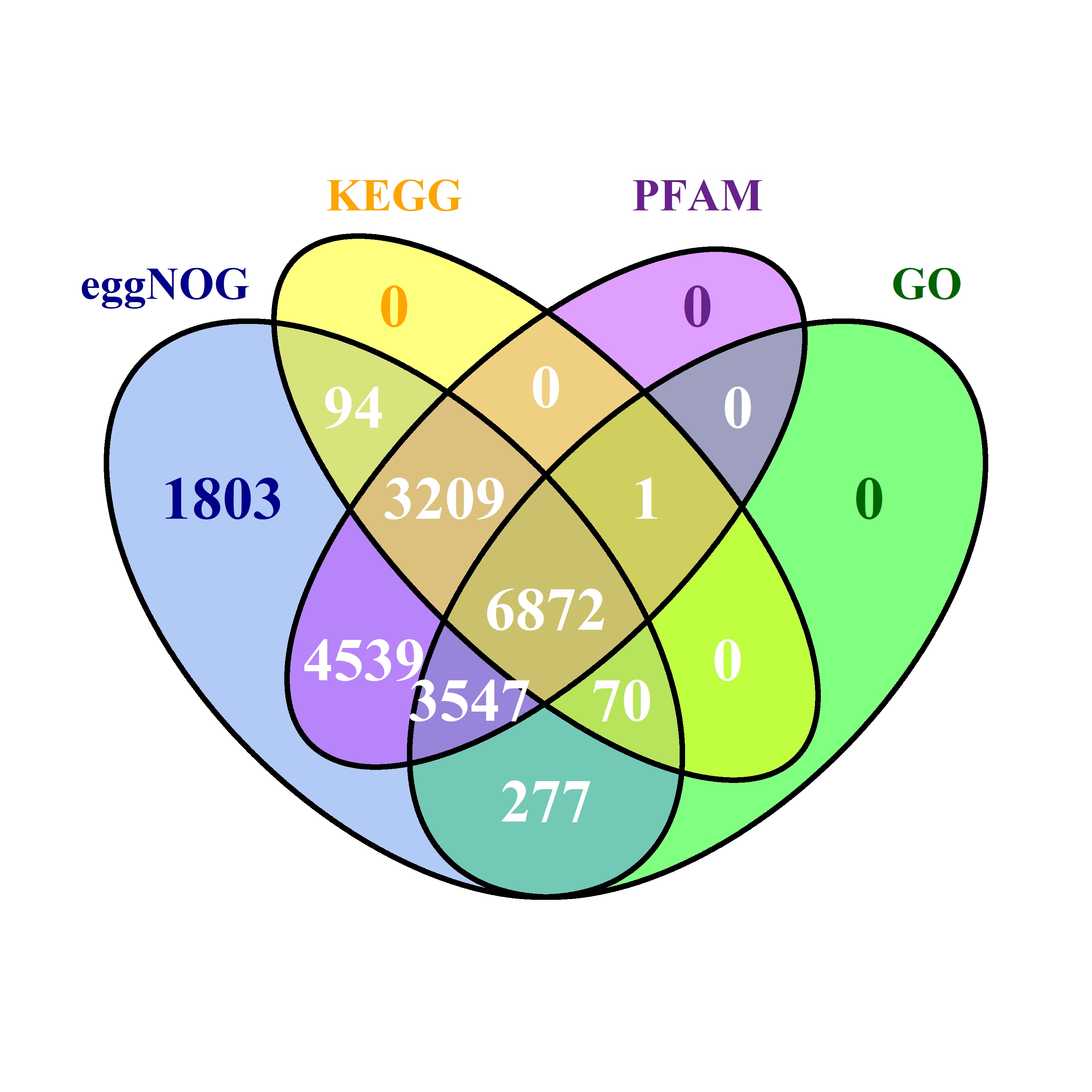
VennDiagram包参数参考VennDiagram: Generate High-Resolution Venn and Euler Plots (r-project.org)
1 | # UpSetR包做upset图 |
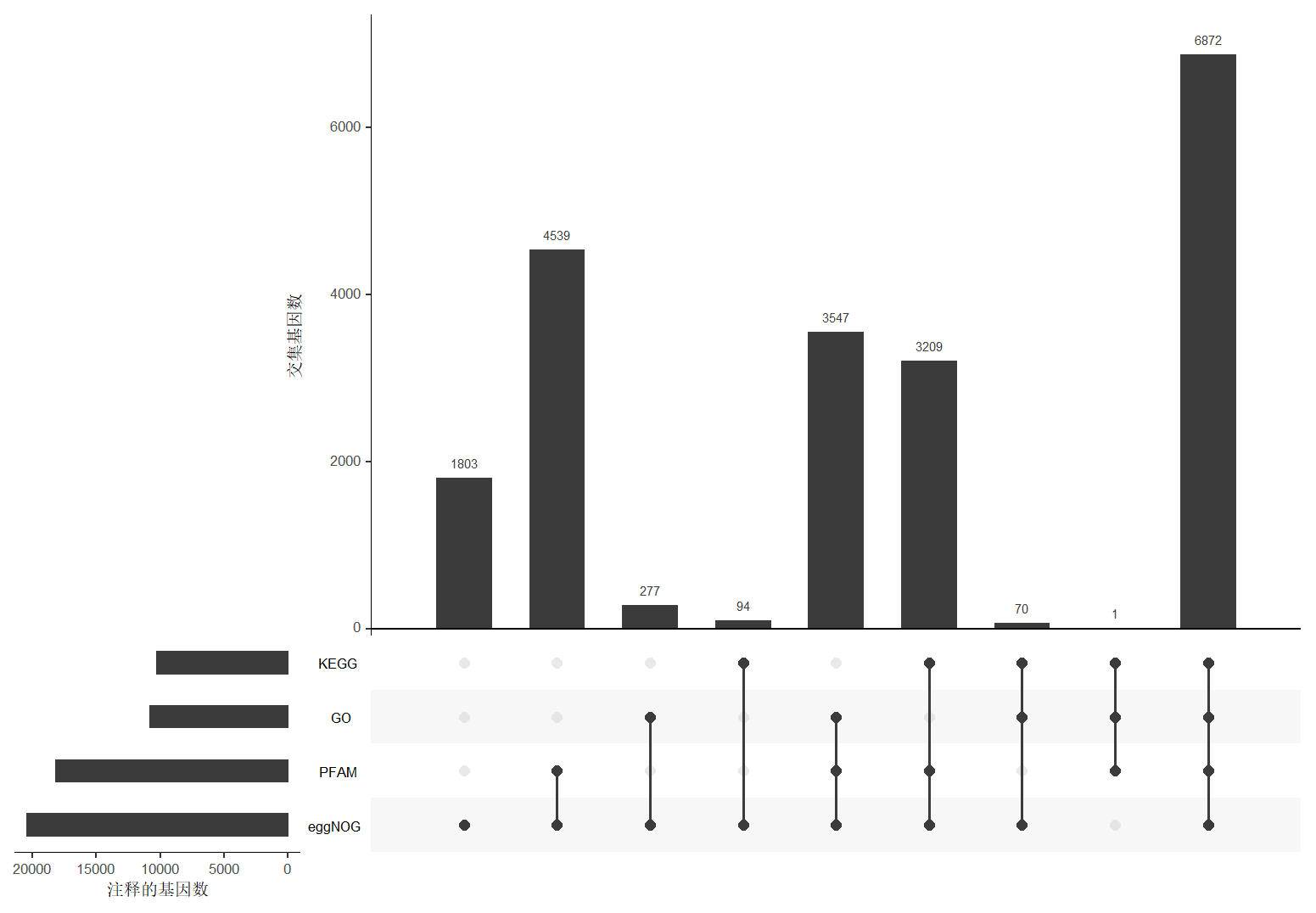
有点扯远了,两个图能获得的信息是一样的(upset图看不懂的话可以对照韦恩图,看柱状图数字以及底下的点和线就明白了),作图不是为了炫技而是让人一目了然,个人感觉upset图更直观一点,当然也可以直接整理一个表格(很多测序公司的报告都喜欢这么做,感觉也没啥意义)。
基于不同数据库得到的基因功能注释结果进行统计,结果显示能注释到功能数据库的基因数目为20411个,占预测基因总数的92.04%,具体如下:
| Type | Number | Percent(%) |
|---|---|---|
| eggNOG | 20411 | 92.04 |
| GO | 10766 | 48.55 |
| KEGG | 10245 | 46.20 |
| Pfam | 18167 | 81.92 |
| Overall | 22176 | 100.00 |
Type: 各数据库类型
Number: 注释的基因数目
Percent: 个数据库注释到的基因所占的预测基因的比例
Overall: 总的预测基因数
2. BUSCO评估
前面的博客写过一篇如何做BUSCO评估——0基础学习基因组三代测序组装(7)——基因组组装质量评估(BUSCO、LAI指数),当时是评估组装的基因组完整性。这里注释完成后也需要对预测的基因集进行评估,和前面不一样的地方是,这里我们用BUSCO的Protein mode。
以前已经下载过真双子叶库的BUSCO保守基因序列了,这里就直接用:
1 | !/bin/bash |
用busco自带的作图脚本处理一下结果文件short_summary.specific.eudicots_odb10.Ap.txt:
1 | mkdir result |
20秒不到就可以出图片结果:
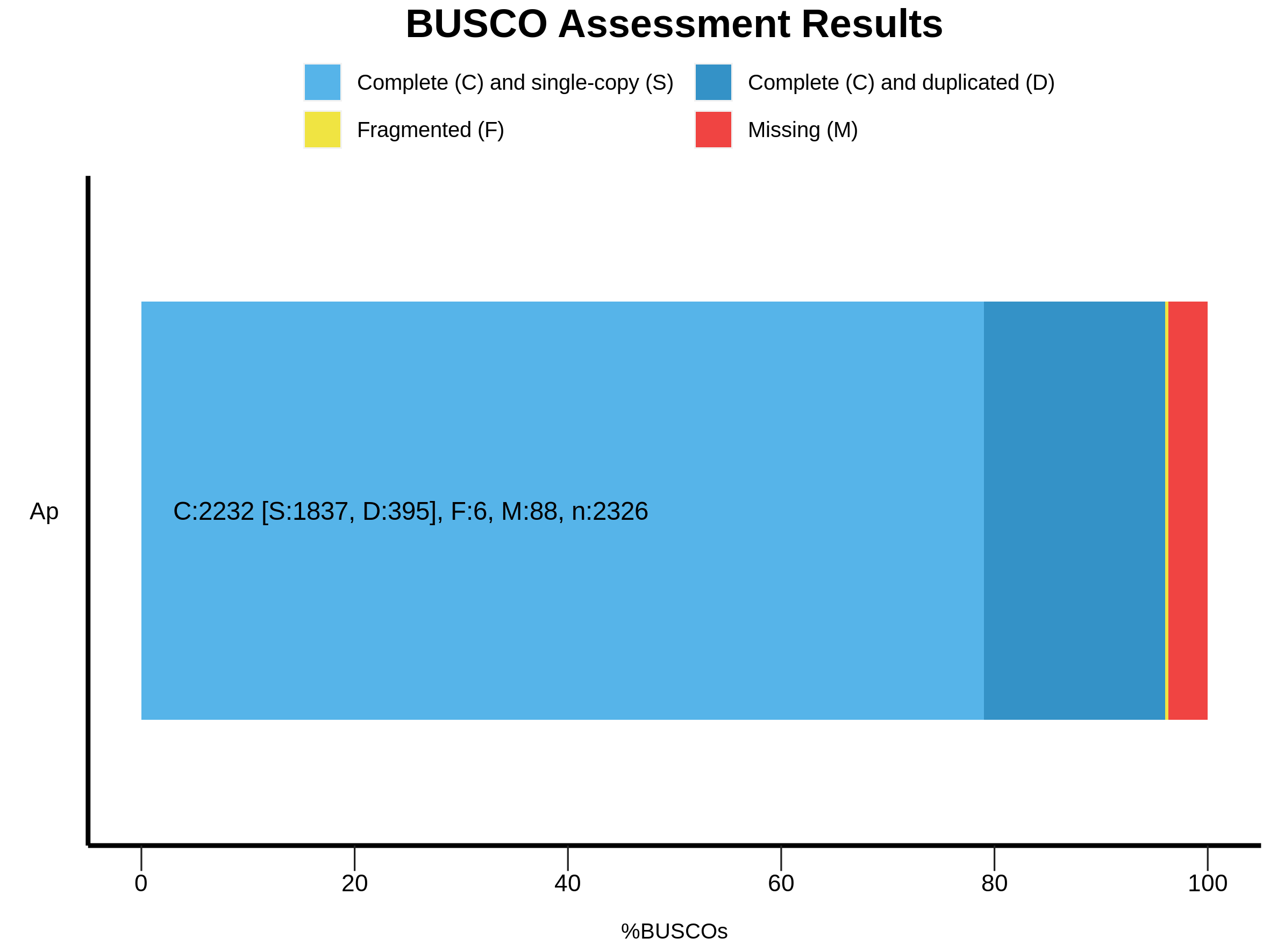
也可以根据结果文件整理一个表格:
| Gene ID | Number | Percent(%) |
|---|---|---|
| Complete BUSCOs(C) | 2232 | 95.96 |
| Fragmented BUSCOs(D) | 6 | 0.26 |
| Missing BUSCOs(M) | 88 | 3.78% |
| eudicots | 2326 | 100.00 |
eudicots_odb10真双子叶库有2326个BUSCO groups,2232个(95.96%)BUSCO groups能够完整比对上(包括1837个单拷贝和395个多拷贝),说明这个基因组注释的完整性还是不错的。
3. 基因转录组表达
用二代转录组数据比对前面组装的参考基因组,统计样本中表达量大于0的基因数。表达量可以用FPKM或者TPM来衡量,最好是用TPM,给定一个标准,比如我这里统计TPM>0.001的基因,就是转录组的一系列标准操作。
简单写个从转录组下机数据比对到定量的脚本,需要新建一个文件夹提交作业:
1 | !/bin/bash |
最终得到result.gtf,这个文件详细记录了每个转录本表达量:
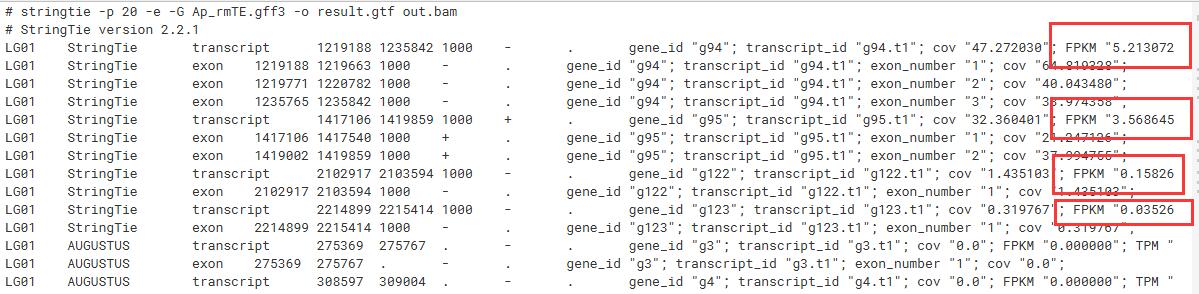
一行显示不下……后面还有个TPM值,简单写个python处理下数据,统计TPM值大于0.001的基因个数:
1 | count = 0 |
也就是说有19760个表达的基因是受转录组数据支持的,占总基因数的89.11%。严格来说这边用的数据是注释功能基因之前的,用到的gff文件是移除TE序列后修改的gff文件,用来评价功能注释的结果似乎没有说服力还不够。这里没有对基因去冗余,计算的表达量和转录组分析的表达量还是有差异的。