



在基因组注释(4)——基因预测这篇博客中记录了怎么用braker3进行蛋白编码基因的预测,当时为了方便安装和使用,直接下载了官方的singularity容器。用过braker3的朋友会发现,官方给的BRAKER标准运行流程中是不包括UTR区域预测的,也就是说,最后得到的gtf/gff文件中没有3’或者5’UTR区域的信息。
前排提示,以下操作是个人尝试,不保证一定正确。
需求描述
当你要克隆某个基因,如果设计的引物不包含UTR区域,会导致pcr出来的基因不完整,为了长远考虑还是要把UTR信息加到注释结果中。怎么才能在braker结果中添加UTR区域的注释呢?
官方注释策略
Braker3官方提供了两种注释策略,其实就是两个参数--UTR=on和--addUTR=on。这两个参数不能同时使用,--UTR=on会根据RNA-seq的数据训练AUGUSTUS的UTR模型,并最终生成包含utr注释信息的gtf文件。看似一步到位,实际上有诸多问题,看官方的issues中很多人提到utr注释信息和基因不匹配,有些utr区域与CDS区域相隔甚远,这明显不符合常理。开发者的回复是使用--UTR=on参数会对UTR进行训练和预测,可能会使UTR注释结果变糟糕,需要谨慎使用:

--addUTR=on参数是在AUGUSTUS预测基因的结果上,直接提取RNAseq比对产生的bam文件中的coverage信息,不进行UTR的训练和预测,直接将UTR加到AUGUSTUS的结果文件中(实现方式是通过开发者编写的软件GUSHR)。需要注意这个参数并不是在第一遍跑BRAKER3的时候加的,我们需要正常流程(不加–UTR=on这个参数)跑一遍,得到结果文件后,再用braker.pl跑一遍下面得命令:
1 | braker.pl --genome=../genome.fa --addUTR=on \ |
刚需是--genome、--bam、--AUGUSTUS_hints_preds和--skipAllTraining这四个参数,augustus.hints.gtf这个文件可以从braker结果文件的AUGUSTUS文件夹中找到。
使用singularity碰到的问题
如果你和我一样用的singularity容器,你会很尴尬的发现加上上面任意一个参数都运行不了,会报错JAVA_PATH无法找到或者其他java相关的问题,我先后遇到了下面的场景:
JAVA error when adding UTR options · Issue #638 · Gaius-Augustus/BRAKER (github.com)
set_JAVA_PATH bug…? · Issue #584 · Gaius-Augustus/BRAKER (github.com)
即使我知道报错的地方想修改braker.pl文件,但是我们这个singularity只提供只读的仓库啊(singularity的缺点又体现了可恶……),我试了3.0.3版本和2.0.2版本都存在这个问题,所以如果用了singularity镜像仓库,就不要想着直接加参数预测UTR区域了……
针对将UTR信息加入到braker.gtf文件的需求,开发者两周前又更新了一个将stringtie组装转录本的中间文件信息加入到braker.gtf文件的脚本:
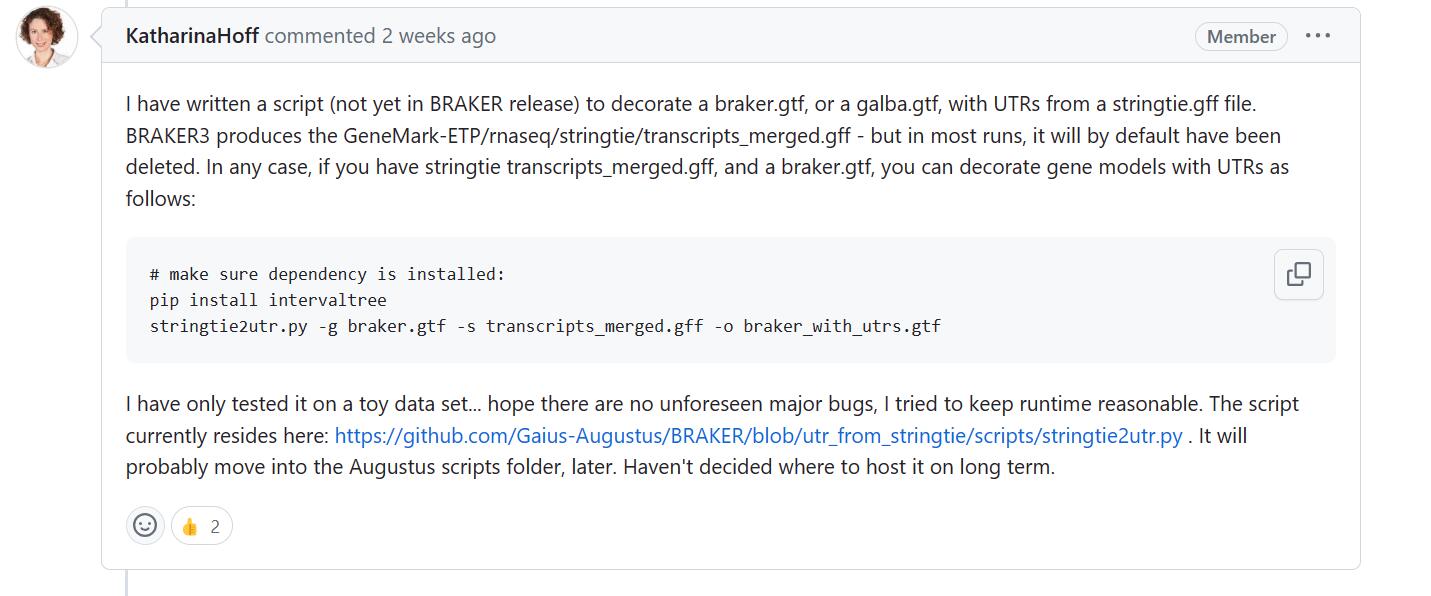
地址如下:
https://github.com/Gaius-Augustus/BRAKER/blob/utr_from_stringtie/scripts/stringtie2utr.py
很可惜的是……transcripts_merged.gff是个中间文件,正常情况下跑完braker就被自动删除了……(你可以在braker的log文件中看到它是啥时候被删的)也不要试图自己再跑一遍stringtie,亲自试了下就算最终得到stringtie的gff文件,跑上面的流程也是跑不通的,得用braker流程的stringtie中间文件(格式不兼容)。
比较坎坷的解决方案
当然也不是完全没办法,用开发者写的软件GUSHR,只需要用hisat将RNA-seq数据比对参考基因组,比对文件转成bam文件后作为输入文件直接用,起到和braker运行加入参数--addUTR=on一样的效果。
1 | #!/bin/bash |
上面的脚本处理转录组下机数据,最终得到RNAseq.bam文件。
接着克隆GUSHR的github镜像仓库:
1 | git clone https://ghproxy.com/https://github.com/Gaius-Augustus/GUSHR.git |
下载gtf2gff.pl:Augustus/scripts/gtf2gff.pl at cc41cd053721756dd6c0aba2f5c306054edd9151 · Gaius-Augustus/Augustus (github.com)
因为不想改环境变量,直接改了下gushr.py的源码,把gtf2gff.pl文件绝对位置写在gtf2gff变量里。
1 | # 197行 |
可以先跑一个test测试一下(需要另外下载bam文件),没有问题就可以带入自己相关的文件跑这个脚本了。
1 | ./gushr.py -t ~/biosoft/braker3/Ap_mydb/Augustus/augustus.hints.gtf -b Ap/RNAseq.bam -g ~/Genome/Ap.fasta -o utrs |
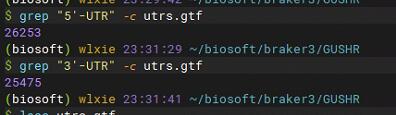
可以看到最后的utrs.gtf文件中注释上了UTR区域(并不是每个基因都能注释到)。
需要注意的是,这里是给AUGUSTUS结果augustus.hints.gtf加上UTR注释信息,而不是braker.gtf(用这个文件跑上面的程序也是会报错的)。开发者对两个gtf有如下的解释:
you have two options:
- augustus.hints.*: This is the final output of AUGUSTUS with protein hints.
- braker.{gtf,gtf3}: This is a union of augustus.hints.* and most reliable genes from GeneMark-EP+ prediction, which is a part of the BRAKER2 pipeline with proteins. Thus, this set is generally more sensitive (more genes correctly predicted) and can be less specific (more false-positive predictions can be present).
- The remaining files are intermediate results that are useful for development and debugging purposes.
The results of augustus.hints.* and braker.{gtf,gtf3} will be quite similar, if you prefer sensitivity, use braker.{gtf,gtf3}. Use augustus.hints.* otherwise.
也就是两个文件差别不大,braker.gtf灵敏度高,但是假阳性多。
也有人提过将braker预测结果直接加入UTR注释(非AUGUSTUS训练UTR模型,因为结果不稳定),开发者提到这并不是一件容易的事,因为braker.gtf格式与UTR预测不兼容……原话如下:
there is currently no easy way to do this because the format of
braker.gtfis not compatible with UTR procedure.How many extra genes do you have in
braker.gtfcompared toaugustus.hints.gtf? In case it’s not many genes, it should be OK to just useaugustus.hints_utr.gtfand add the extra genes frombraker.gtfto the result (the extra genes will haveGeneMark.hmmin the second column).
然而这两个文件的基因和转录本编号并不是一一对应的……直接将braker.gtf第二列GeneMark.hmm加到刚刚UTR注释的augustus.hints.gtf中会导致基因和转录本id混杂,并且有些AUGUSTUS预测的基因位置会和GeneMark标的基因位置重复。
权衡了一下,还是直接用augustus.hints.gtf作为结果文件比较合适,经过上面的UTR注释后,这个gtf文件整体会小一圈,因为删除了intron和exon信息,只保留了CDS、start_codon、stop_codon、transcript、gene以及5‘和3’-UTR。
别忘了这个gtf是非标准的gtf文件,还需要经过两步处理…..(真的头疼)
1 | # 转换该gtf文件为gff3文件 |
perl文件来自于braker singularity容器的/usr/share/augustus/scripts/gtf2gff.pl。或者可以到AUGUSTUS官方仓库取:
这个是开发者写的gtf转gff3脚本,然而转了以后还存在空格问题,无法被常规的gff软件解析,如下:
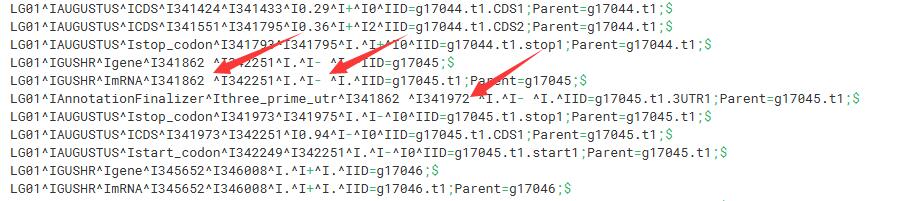
需要自己再处理一下:
1 | # 将含有AnnotationFinalizer和GUSHR字段的行提取,去除空格 |
最后的问题
最后这个final.gff3文件表面风平浪静,实际还存在一些问题需要手动排查:
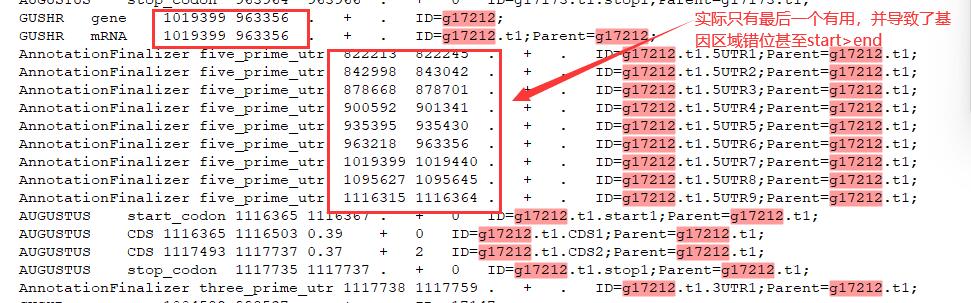
我跑了一遍有三十多个基因存在start位置大于end位置的情况,这可以手动修改。但是预测的5’和3’UTR区域不止一个,有的也会相隔甚远,基本上来说,只有最后一个5’UTR区域和第一个3’UTR区域比较靠谱,所以仍然要写脚本筛选UTR,并修改gene和mRNA所在的位置。
这种问题就要扒gushr.py的源码看作者对UTR区域筛选的处理方式了……emmmmm还是直接说结论吧,braker的结果对UTR区域的注释确实不那么友好,现阶段我只能给出上面的思路,最后还是要写脚本自己处理gff文件筛选UTR区域,以后有空再去实现了。
现在比较着急想要某基因的CDS上下游区域怎么办呢?
从注释文件中查找到目的基因,拿gff文件和基因组文件,直接提取braker预测的基因上下游一段区域,自己取舍一下。
有没有更好的方式注释UTR区域呢?
我的理解是最好测一个全长转录组,以组装的基因组做参考基因组,拼接非冗余的全长转录本,可以比较明确的区分各基因的UTR区域。最后记得导入UCSC基因组浏览器看一看结果。
总之这么长一串的踩坑和处理记录,估计别人也都用不了,就当是自己操作的备份吧hhhhhhhhh