



有的时候存在这种情况:我手上有两个近缘植物的基因组测序数据,这两个物种可能没有人做过,或者别人做过但是没有提供参考基因组。而课题组因为经费不足,只测了一个物种的Hi-C(嗯,说的就是我),那如何以组装的基因组为参考,把另一个近缘物种基因组也组装到染色体级别呢?
记录一下基于近缘物种参考基因组的染色体水平组装和注释,用到的软件是RagTag以及配套的Liftoff。
1. RagTag
RagTag是一个纯python编写的软件工具集(但并不是所有功能都是python实现的,比如minimap2/Nucmer/unimap是通过subprocess模块调用命令行使用,产生子进程实现的),用于将组装的contig级别的基因组提高到染色体级别。具体来说可以做到以下三个功能:
- 基于同源的组装错误纠正
- 基于同源的scaffold组装以及修补(也就是填补gap)
- scaffold合并(不同方式得到的scaffold合并)
同时官方提供了处理常见基因组组装文件格式的命令行实用程序,也是纯python编写的,都在file utilities文件夹中,主要是实现以下几种功能:
agp2fa: 将AGP文件转换成fasta文件。AGP文件是描述contig如何构建成scaffold的,可以看NCBI对该文件类型的描述:AGP Specification v2.1 (nih.gov)agpcheck: 验证AGP格式是否正确asmstats: 统计assembly序列的信息splitasm: 按照gap分隔assembly序列,为后续处理提供AGP文件delta2paf: 转化delta文件到PAF文件。delta文件是NUCmer基因组比对软件(NUCleotide MUMmer)产生的结果文件,作用是记录每个联配的坐标,每个联配中的插入和缺失的距离。PAF文件也是类似描述两组序列之间近似的联配位置的文件,是minimap2的输出文件。点击蓝色字体可以看两种格式的具体样式。paf2delta: 和上面相反updategff: 根据RagTag的AGP文件更新gff文件(不是得到最终基因组的gff文件,要想得到基因组的gff文件,作者推荐用配套的软件Liftoff)
RagTag运行流程主要分4步:

官方仓库:RagTag/ragtag_correct.py at master · malonge/RagTag (github.com)
1.1 correct
RagTag的校正模块,使用参考基因组来识别和校正基因组中潜在的错误组装。这一步直会将可能存在错误组装的序列打断,不会增加或者减少原序列大小。
1 | usage: ragtag.py correct <reference.fa> <query.fa> |
详细参数-h可以查看,需要注意reference.fa是参考基因组(同一个物种或者近缘物种),query.fa是我们需要组装的contigs(contigs是通过二代+三代测序下机数据组装的)。
1 |
|
得到的结果文件:
├── ragtag_output
│ ├── ragtag.correct.agp
│ ├── ragtag.correct.asm.paf
│ ├── ragtag.correct.asm.paf.log
│ ├── ragtag.correct.err
│ ├── ragtag.correct.fasta
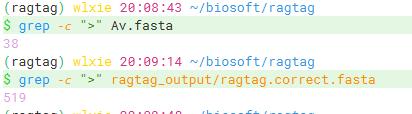
可以看到contigs从原来的38条被切断成了519条:
1.2 scaffold
RagTag的脚手架(scaffold)组装模块,将组装的草稿(draft assembly)序列排序和重定向为更长的序列。使用上一步纠错后的contigs比对参考基因组序列,contigs之间的gap使用N(默认100个)填充,同样这一步不会改变原来的序列。
1 | usage: ragtag.py scaffold <reference.fa> <query.fa> |
详细参数-h可以查看,reference.fa是参考基因组,query.fa是上一步纠错后的序列。
1 |
|
-C这个参数可以把没有比对上的序列都放在chr0这条假想的染色体上。
得到的结果文件:
├── ragtag_output
│ ├── ragtag.scaffold.agp
│ ├── ragtag.scaffold.asm.paf
│ ├── ragtag.scaffold.asm.paf.log
│ ├── ragtag.scaffold.confidence.txt
│ ├── ragtag.scaffold.err
│ ├── ragtag.scaffold.fasta
│ └── ragtag.scaffold.stats
统计下各scaffold序列的长度:
1 | $ seqkit fx2tab --length --name --header-line ragtag_output/ragtag.scaffold.fasta |
因为我的参考基因组是11条染色体,这里大部分contig都能比对上,长度也正常,剩下的contig都在Chr0_RagTag这条序列。
ragtag.scaffold.stats这个文件可以查看比对上scaffold上的contig信息:
1 | $ cat ragtag.scaffold.stats | column -t -s $'\t' |
第一步拆分的519条contigs有90条可以比对上,429条未比对上参考基因组,但是这429条序列总长度却只有5721732 bp。根据上面的信息也可以知道,产生的507个gap中有428个在Chr0_RagTag这条序列中(unplaced_sequences都在这),真正在染色体上的gap数量是79,可以写个脚本统计一下各条染色体上gap数量:
1 | # count_gap.py 作用是统计这一步产生的各scaffold上的gap数量 |
可以对上gap数量,没有问题。
1.3 patch
RagTag的填补模块,关于这个模块,国内的各方帖子都说是填补上一步骤产生的有gap的scaffold序列。对,但是不完全对。patch和scaffold是两个独立的模块,且两者都可以独立完成scaffolding的过程。
scaffold模块中,我们以参考基因组序列为基准,将contig定向和排序成为scaffold,整个过程没有发生contig序列的增加或者减少。
patch模块和gap填补软件类似,一般情况下是用三代长读长序列(ONT Ultra-long reads这种)填补gap,但是这里用的是assembly序列,且填补前后序列会发生变化。patch模块有两种运行模式:
--fill-only只填补已存在的gap,就和传统的gap填补工具类似
--join-only不填补已存在的gap, 会对contig重新进行定向和排序,这会产生新的gap,且填补的是新产生的gap
fill模式的原理如下:
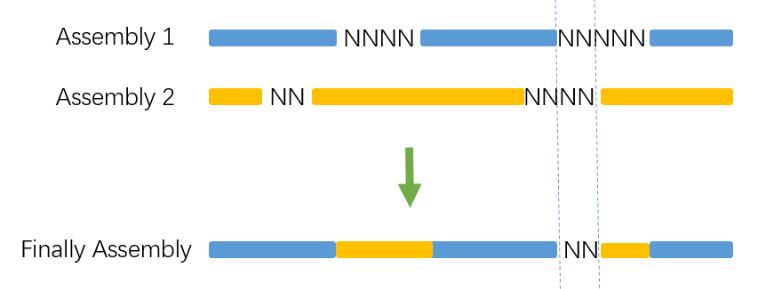
默认情况下,两种运行模式都会进行,可以看到填补的基础是需要另一条同一个物种的assembly序列。
如果你前一步跑了scaffold模块,仅仅只需要填补该过程产生的gap,那麽只要以--fill-only的模式运行patch模块即可。
因为我没有其他assembly序列,所以跑scaffold模块就够了,为了测试一下这个模块,我又用原始assembly序列跑了一遍:
1 | usage: ragtag.py patch <target.fa> <query.fa> |
详细参数-h可以查看,这里target.fa是上一步产生的scaffold序列,query.fa是我一开始跑correct模块的原始assembly序列。
1 |
|
得到的结果文件:
├── ragtag_output
│ ├── ragtag.patch.agp
│ ├── ragtag.patch.asm.delta
│ ├── ragtag.patch.asm.delta.log
│ ├── ragtag.patch.asm.paf
│ ├── ragtag.patch.comps.fasta
│ ├── ragtag.patch.comps.fasta.fai
│ ├── ragtag.patch.ctg.agp
│ ├── ragtag.patch.ctg.fasta
│ ├── ragtag.patch.ctg.fasta.fai
│ ├── ragtag.patch.err
│ ├── ragtag.patch.fasta
│ ├── ragtag.patch.rename.agp
│ ├── ragtag.patch.rename.fasta
│ ├── ragtag.patch.rename.fasta.fai
这里结果文件较多,有必要说一下几个文件的作用(来自官网):
file name Description ragtag.patch.agpThe final AGP file defining how ragtag.patch.fastais builtragtag.patch.asm.*Assembly alignment files ragtag.patch.comps.fastaThe split target assembly and the renamed query assembly combined into one FASTA file. This file contains all components in ragtag.patch.agpragtag.patch.ctg.agpAn AGP file defining how the target assembly was split at gaps ragtag.patch.ctg.fastaThe target assembly split at gaps ragtag.patch.errStandard error logging for all external RagTag commands ragtag.patch.fastaThe final FASTA file containing the patched assembly ragtag.patch.rename.agpAn AGP file defining the new names for query sequences ragtag.patch.rename.fastaA FASTA file with the original query sequence, but with new names
我们关注的是最后的结果文件ragtag.patch.fasta。这一步运行的时间很长,多长呢?和前面两个步骤放一起感受一下:
1 | $ sacct --format=JobName,Start,End,Elapsed,NCPUS |
前两步分分钟完成,patch这一步需要花2小时。
还有一点需要注意,如果用来填补gap的序列是T2T或者近似T2T的序列,需要考虑到它们包含大量的高度重复的序列,RagTag的pactch模块是通过query contig和至少两个target contig之间的唯一比对(unique alignments),来确定潜在的填补区域。大量的重复区域会导致唯一比对出现大量的gap,从而误判这个区域不是潜在的填补区域。
作者提出的建议是加入参数-i并调整这个值,来控制最大对齐中断长度(maximum alignment break length),可以更大限度容忍唯一比对中出现的长gap区域。或者直接将query序列重复区域打断(也就消除了重复序列导致的非唯一比对)。
看了下结果文件,几乎和scaffold模块一样,就填补了几个gap(因为我没有其他assembly序列),而且对Chr0_RagTag这条多余contig合在一起的序列,填补gap完全没意义,于我而言这步结果意义不大且更不可信,只是为了测试这个模块。主要还是用上一步的ragtag.scaffold.fasta文件。
1.4 merge
RagTag的合并scaffolding结果的模块,起到改进scaffolding结果的作用。
因为scaffolding过程可以使用多种方式,比如使用Hi-C技术、Bionano的光学图谱技术、遗传图谱等等,而merge模块的作用是将各种技术的scaffolding结果(AGP格式)合并到一起,提高基因组组装草图的精度。
这一步我没有Hi-C数据做测试了,就放一下官方的使用方法,用到再做详解:
1 | usage: ragtag.py merge <asm.fa> <scf1.agp> <scf2.agp> [...] |
2. Liftoff
Liftoff软件的作用是将同一或者近缘物种的基因组注释映射到组装的基因组中。输入文件很简单,一个组装的基因组,一个参考基因组以及注释文件即可。
这款软件是使用Minimap2将基因序列从一个基因组比对到另一个基因组(不是基因组之间的比对),每个基因都会寻找外显子的比对,以最大限度提高序列的同一性,同时保留转录本和基因结构。如果两个基因都比对到overlapping位置,还会判断哪个基因最有可能是错误比对,并进行重新比对(挺好奇怎么实现的?)。
官方仓库:agshumate/Liftoff: An accurate GFF3/GTF lift over pipeline (github.com)
1 | usage: liftoff [-h] (-g GFF | -db DB) [-o FILE] [-u FILE] [-exclude_partial] |
详细参数-h可以查看,跑一个流程试试:
1 |
|
两分钟就可以跑完,结果文件如下:
├── Av.gff3
├── intermediate_files
│ ├── reference_all_genes.fa
│ └── reference_all_to_target_all.sam
└── unmapped_features.txt
gff3文件是我们要的结果,intermediate_files文件夹中放的是中间文件,没啥用;unmapped_features.txt记录的是没比对上的基因。
提取一下CDS序列和蛋白序列:
1 | $ gffread Av.gff3 -g ragtag.scaffold.fasta -x Av.codingseq |