



snakemake是一款强大的工作流管理工具,用于构建和运行复杂的数据分析工作流。其工作流是基于python语言描述的,类似于Makefile的工作流描述语言。Makefile指定的是源文件之间的依赖关系,以及如何将它们编译成可执行文件和库,而snakemake指定的是数据处理过程中的依赖关系和规则,并自动化执行这些规则生成最终的输出。
Snakemake
snakemake的一些特点:
- 声明式工作流描述: 使用Python风格的语法编写工作流规则,描述数据处理步骤和依赖关系。也可以在Snakefile中直接编写python代码,解决复杂的功能需求。
- 自动化任务调度: Snakemake可以自动解决任务之间的依赖关系,并根据需要并行运行任务,以最大程度地提高效率。
- 灵活性: Snakemake支持复杂的工作流设计,包括并行处理、条件执行、动态文件生成等功能,使用户能够灵活地定制数据处理流程。
- 集成性: Snakemake可以与常见的集群调度系统(如SLURM)集成,方便在计算集群上运行工作流。
- 报告生成: Snakemake可以生成详细的报告,展示工作流执行的结果和性能指标,有助于用户进行结果分析和优化。
官方给出的文档中有非常详细的操作,包括如何安装,以及如何执行和定义一个工作流。官方还有个代码库,存放了大量的生信分析的工作流代码和文件供我们参考。
官方文档:Snakemake | Snakemake 8.10.6 documentation
工作流库:Snakemake-Workflows (github.com),Snakemake workflow catalog
这里记录下我对snakemake的学习过程,以官方文档的例子为主初步理解和学习这个软件的基本用法,再用自己的服务器跑一跑简单的流程。
1. 安装snakemake和下载示例数据
官方推荐使用Conda/Mamba进行安装,因为这两个包管理软件可以很好处理snakemake的软件依赖关系。也可以使用python的pip工具安装,但需要手动解决一些依赖问题。
由于学校集群至今没能联网(对学校这方面的管理非常非常不满,一个集群没有专人管理,出问题找不到管理员解决,也没有用户手册,没有后台监测,导致一些用户乱用登录节点资源。真的推荐向华农学习学习,南疆最大的集群管理如此混乱,说出去真的不好意思),源码安装需要解决的依赖太多,所以暂时不做集群中的演示了,以我的服务器为例跑一跑流程。
1 | # 先安装mamba(base环境下) |
官方的示例是跑一个bwa比对流程,用samtools转sam文件并排序后,最终使用bcftools进行变异检测,一套标准的call genomic variants流程。
示例数据结构如下:
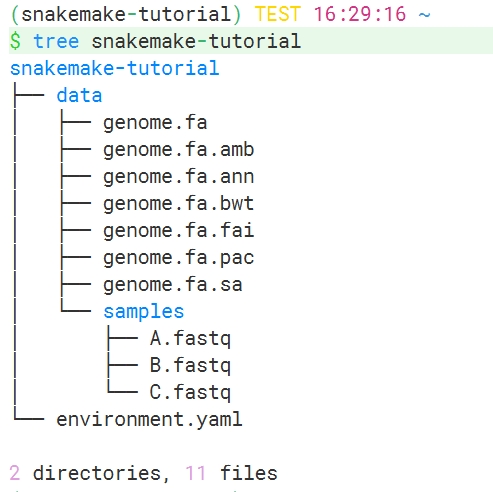
data文件夹中是工作流需要用到的数据,包括基因组序列.fa及其索引文件,和三个测序数据.fastq文件。
environment.yaml文件是示例中需要用到的软件,我这里pip部分需要改源和手动安装(默认的pip源安装软件会超时报错)。
1 | pip install -i https://pypi.tuna.tsinghua.edu.cn/simple pysam==0.22 |
2. snakemake基本用法
基本用法主要参考官方:Basics: An example workflow | Snakemake 8.10.6 documentation
Snakefile是snakemake默认读取的文件,记录程序运行过程中的数据来源、程序命令、参数、输出目录等等。运行snakemake会自动寻找目录下是否有这个文件,如果你改了这个文件的名字,需要在运行时指定参数 -s。
1 | # 创建Snakefile |
以上就是snakemake最简单的用法,以下是使用的两个基本点:
- Snakefile基本组成单位是
rule,定义一个规则,也就是一个处理步骤。 - 每个
rule包含三个基本元素:input、output、shell/run/script。input为输入文件,一个文件一行,注意逗号不能省略,python会连接后续字符串,没有逗号会导致文件读取错误。output定义输出文件,不存在的路径会自动创建。shell/run/script是要在shell中执行的命令,或者要执行的python代码,shell类似于python的subprocess模块创建子进程。
以上Snakefile文件仅支持空格whitespace,不支持缩进写法,tab键无法被识别。当输入输出文件有多行的时候,逗号会将每个文件以一个空格的形式分隔,也就是执行前会将{input}部分替换为data/genome.fa data/samples/A.fastq。
但是shell中不用逗号,可以将两行字符串直接连接,注意每个字符串需要有一个尾随空格(默认是直接连接一起的,两个字符串之间没有空格)。
在运行snakemake前,我们可以伪执行一次检测是否有输入输出文件的错误:
1 | snakemake -np |
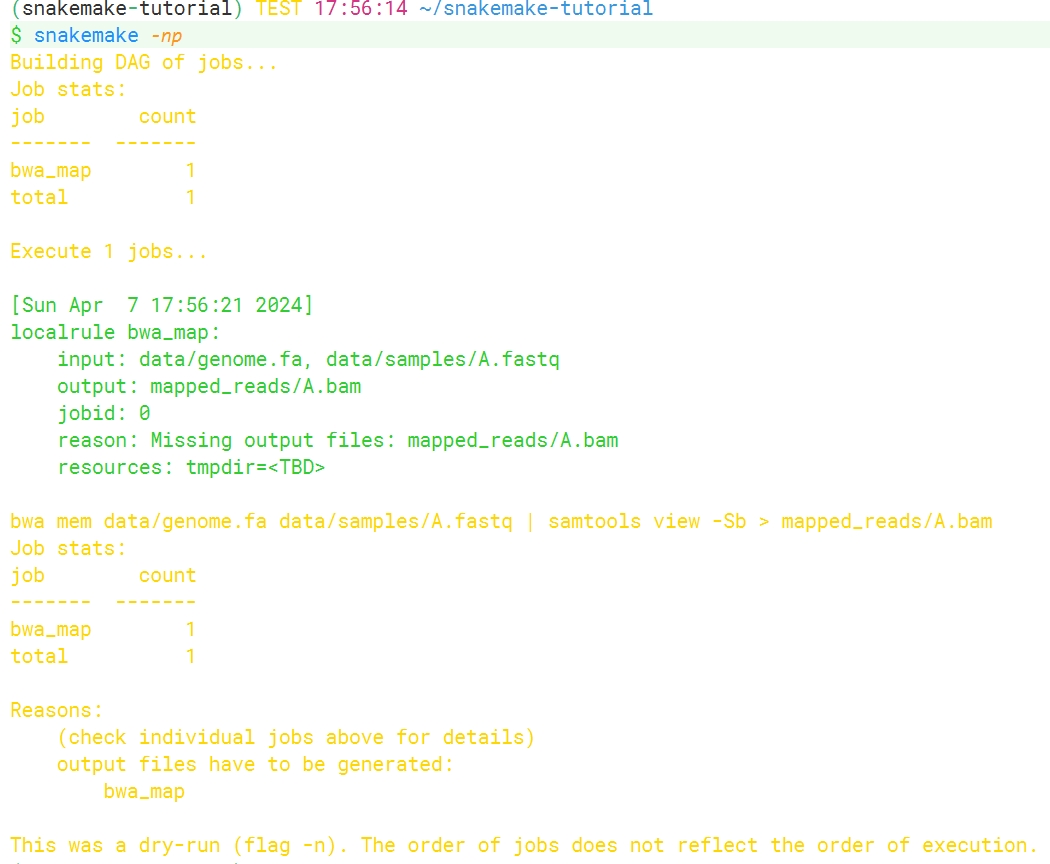
没有报错说明这个rule的逻辑没有问题,可以直接执行:
1 | snakemake --core 1 |
运行时需要指定整个流程同时最多使用的核心数量,每个rule中可以用threads定义该rule使用的线程数。
Snakemake运行的逻辑是指定输出的文件或者目录,Snakemake为了拿到这个结果文件会一层层往上寻找得到该文件的流程,跳过不需要执行的rule。所以书写的时候每个rule之间的顺序不重要,只是书写代码的时候方便我们阅读。
上面的作业命令在运行结束后,结果文件不会被再次创建(即使你重新运行一遍命令),除非更改文件的时间戳,snakemake仅在输入文件比输出文件更新,或者输入文件被其他作业更改后才可以重新运行作业命令。
3. rule all
实际上一个工作流往往含有多个rule,我们会制定多个rule将工作流中的所有步骤模块化写入,snakemake会自动分析不同rule之间的input和output的依赖关系,从而将这个流程串联起来。
rule all是一个特殊的规则,用于定义工作流中的主要输出文件或目标,也就是指定我们希望整个工作流生成的结果。Snakemake 将根据 rule all中定义的目标来确定需要运行的规则,以确保生成这些目标文件,简化工作流的管理和执行过程。
比如上面的例子,我们希望最终得到mapped_reads/A.bam这个结果,可以加上一个rule all:
1 | rule all: |
在复杂一些的流程中会加入这个规则,作为snakemake运行的入口,一般只需要定义input,因为一个流程的最终输出结果是其他rule的output。snakemake会从rule all的input向上一层层回溯,找到最初rule的input文件,也就是我们的原始文件,比如这里的测序文件和基因组文件,从而理清整个流程并依次执行。
如果一个rule的input和output文件不被其他rule依赖,则这个rule不会被执行。
上面的这些操作如果没有在命令行中指定目标文件,且Snakefile中没有定义rule all,将会默认执行第一条规则。
4. 通配符
以上例子我们用了具体的文件名作为输入和输出,实际应用时往往有批量的文件需要作为输入或者输出文件,snakemake可以使用通配符进行命名。
1 | SAMPLES = ['A', 'B', 'C'] |
以上例子就很形象地体现了snakemake的python特性:
expand()是snakemake内部函数,用于定义规则的输入、输出或其他需要动态生成文件路径的地方。{sample}通配符用来指定由SAMPLES列表生成的文件路径。- 这里的
input和output我们可以看做是python中的字典类型变量,比如input.REF表示访问字典input中的键为REF的值,相当于调用了input这个对象的REF属性,当然也就可以写成input[REF],这样可以有效区分输入输出的文件及顺序。
当然,说到python也可以用列表推导式来表示输出的文件名:
1 | # 使用列表推导式,省去了SAMPLES列表。替代rule all规则中input对象expand()函数表示的文件名。 |
如果有多个参数列表,expand()函数也可以对应传入:
1 | expand("mapped_reads/{sample}.{replicate}.bam", sample=SAMPLES, replicate=[0,1]) |
如果样本数多,有一定的规律可循,则可以使用glob_wildcards()函数和使用wildcards对象提取文件名称和路径,这两者都是使用类似shell命令中的通配符访问对象,但是两者在使用方法上有一些区别。
假设我们现在有以下sample的双端测序文件:
1 | files = ["fastq/sample1_R1.fastq", "fastq/sample1_R2.fastq", "fastq/sample2_R1.fastq", "fastq/sample2_R2.fastq"] |
glob_wildcards()可以接受多个通配符表达式,返回的是一个元组类型数据:
1 | (SAMPLES, PAIR) = glob_wildcards("fastq/{sample}_{pair}.fastq") |
wildcards 也就是通配符对象,用来捕获单个文件名中的通配符信息:
1 | (SAMPLES, PAIR) = glob_wildcards("fastq/{sample}_{pair}.fastq") |
shell中也可以直接用三引号将要运行的shell程序包含进去,shell中不能直接识别通配符{sample}和{pair}。在Snakemake规则中,input和 output可以直接使用通配符(wildcards)的语法,如{sample} 和 {pair},来引用通配符的值。在 shell部分中,需要使用{wildcards.<通配符名>} 的语法来引用通配符的值,也就是通过wildcards对象访问其属性值。
回到一开始下载的数据,再次提醒glob_wildcards()函数返回的是元组类型,所以当只匹配一个变量的时候,需要添加逗号!否则会找不到文件:
1 | (SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq") |
顺便再提一句,snakemake在格式化shell命令时,使用花括号{}表示通配符引用的内容,如果在shell命令中有使用花括号的命令,需要使用两个花括号进行转义。
1 | rule example_rule: |
5. 配置文件
一般在snakefile文件中,我们会先定义各种变量名,但是当原始数据比较多的时候全都写在一个文件中会比较乱。为了使snakemake脚本更加通用,可以使用配置文件,将分析需要用到的原始数据、参考基因组等写入到配置文件中。
配置文件可以书写成JSON或者YAML格式,读入为python的字典类型对配置参数和值进行定义。
以官方代码仓库中的call SNP流程的部分配置文件为例:
1 | samples: config/samples.tsv |
在snakefile中调用配置文件参数:
1 | # common.smk定义了各种需要用的脚本以及配置参数 |
可以看到调用字典key值的方式就可以定义配置参数了,用configfile: "config.yaml"读取成字典,变量名为config。
这个例子定义了很多子模块执行不同的功能,就像搭积木一样,将这些模块搭成一套完整的分析流程。
6. 其他常用关键字
上面的例子中还有几个其他常用参数顺便也记录下。
6.1 日志 log
在运行的时候我们会发现snakemake的日志是直接输出到屏幕的,我们可以选择保存的位置,使用log参数定义输出的日志。运行出错时,在log里面定义的文件不会被snakemake删掉,而output里面的文件则会被删除。
6.2 规则参数 params
有时在shell中,需要根据不同的样本使用不同的参数,该参数其既不是输入文件,也不是输出文件,如bwa mem -R 参数。如果把这些参数放在input里,则会因为找不到文件而出错。snakemake使用params关键字来设置这些参数。
bwa mem -R 设置reads标头,放到一对引号中,也就是sam文件中的RG部分,同一样品可能包括多个测序结果,来自不同lane,不同文库,或者不同样品的比对结果合并到同一个文件中进行处理,需要通过RG进行标记区分。RG每个标记用冒号分割键和值,不同标记用 ‘\t’ 分隔。例如‘@RG\tID:foo\tSM:bar\tLB:library1’
1 | (SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq") |
6.3 运行时间和内存
在该关键字下的文件会自动记录该规则运行所消耗的时间和内存。
1 | (SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq") |
6.4 wrapper
指定一个包装器(wrapper)脚本,这个包装器脚本可以用来自动化地配置和运行特定软件或工具。使用 wrapper 关键字可以简化规则的编写,尤其是对于需要复杂参数设置或环境配置的软件工具。
具体来说,wrapper 关键字的作用包括:
- 自动化软件配置:通过指定一个包装器脚本,可以在规则中自动化配置软件的安装路径、参数设置、环境变量等。这样可以简化规则的编写,并确保软件的正确配置。
- 版本控制:包装器脚本通常会包含特定软件版本的配置信息,这样可以确保在不同环境中使用相同版本的软件,避免版本不一致导致的问题。
- 参数设置:包装器脚本可以预先设置好软件的参数,使得在规则中调用软件时不需要重复指定参数,减少了重复劳动。
- 环境隔离:包装器脚本可以帮助实现软件运行环境的隔离,确保规则中调用的软件在一个独立的环境中执行,避免与其他软件或库的冲突。
比如前面的例子用了"0.74.0/bio/reference/ensembl-sequence"这个wrapper值,实际上用的是以下包装好的脚本:
ENSEMBL-SEQUENCE | Snakemake wrappers (snakemake-wrappers.readthedocs.io)
这些打包好的脚本可以在如下的官方网站查找:
The Snakemake Wrappers repository | Snakemake wrappers (snakemake-wrappers.readthedocs.io)
6.5 其他
不一一列举了,放个表格:
| 关键字 | 描述 | 关键字 | 描述 |
|---|---|---|---|
| message | 打印消息 | threads | 定义该规则线程数 |
| resources | 定义使用的资源,如内存、CPU | version | 定义规则版本 |
| conda | 定义conda环境 | singularity | 容器环境运行 |
| run | 定义多行命令 | shell | 执行shell命令 |
| script | 定义规则中处理脚本 | notebook | 执行jupyter notebook文件 |
运行参数:
1 | # 设置出错后重新运行次数 |
生成的有向无环图(也就是这里的流程图)如下:
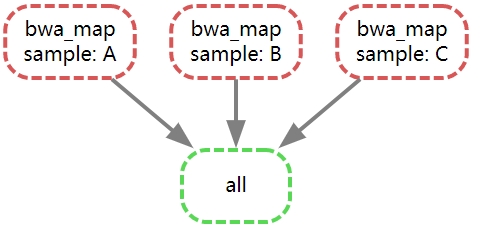
总结
自己只是简单粗浅地了解了下Snakemake这个工作流软件,越是了解越是发现其便捷和强大之处。之前做生信分析流程东一块代码西一串数据,写的博客也是零零散散,真的很有必要把流程给串起来形成一套方便维护的流程代码,需要学习的东西还有很多。
Snakemake还有非常多的优点这里没有一一体现出来,比如其支持大规模的平行计算(等到学校集群联网了,一定试试怎么投递slurm作业),可移植性强(多种依赖工具的支持,conda环境、singularity、jupyter),分析流程模块化(分解成多个rule,用include调用)。
自己在摸索的时候也看到官方很多工作流示例,国内也有很多质量非常高的进阶教程,都需要我慢慢去理解和动手测试。
Snakemake+RMarkdown定制你的分析流程和报告-腾讯云开发者社区-腾讯云 (tencent.com)
这里先挖个坑,有空把我使用GATK call SNP和INDEL的流程自己写一遍,加深对snakemake的理解,再请教下师兄怎么联合jupter使用snakemake,毕竟写python代码还是jupter用起来比较舒服,测试也比较方便。