



最近基因家族分析的文章越来越多(铺天盖地的培训班宣传),知网上几乎天天都有更新这类文章。很多的这类文章都是纯生信分析,用的公共数据库中的基因组、蛋白序列和转录组数据,最多加个qPCR验证(甚至有些文章都没有),内容没有深度比较氵。写这篇笔记并不是鼓励大家水文章,只是掌握一门分析方法,不要把这类分析看得太难,0代码基础也可以做分析。
开始前需要准备以下数据:
- 待分析物种蛋白序列(基因组或者转录组得到的都行),鉴定基因家族用
- 基因组序列及注释文件(
gff或者gtf都可),鉴定基因家族的染色体分布、结构分析、共线性分析以及启动子区域顺式作用元件预测用 - 模式植物的待分析基因家族蛋白序列(比如拟南芥),构建进化树用
鉴定基因家族
基因家族(gene family),是来源于同一个祖先,由一个基因通过基因重复而产生的一组基因,它们在结构和功能上具有明显的相似性,编码相似的蛋白质产物,行使相似的功能。同一个基因家族有着相同的保守结构域(domain),我们可以根据这个特点从蛋白序列中筛选目标基因家族成员。
以我做的WRKY基因家族为例,简单讲讲怎么鉴定候选的基因家族成员,这里只需要用到待分析物种的蛋白序列即可。
1. 通过注释获得候选基因家族
将蛋白序列(数量要少于10万条)先在直系同源蛋白注释网站eggNOG-mapper上自动注释一遍:
这个网站的用法在前面的博客中有写,不罗嗦了。基因组注释(6)——在线版eggNOG-mapper注释功能基因
目的是为了获得结果文件out.emapper.annotations.xls最后一列Pfam数据库的注释(其他列的KEGG和GO注释结果也可以用来做富集分析)。Pfam数据库是个蛋白家族数据库,提供各个基因家族的隐马尔可夫模型(HMM)。
网页版eggNOG-mapper的注释原理就是用hmmer3工具将所有蛋白序列在Pfam数据库搜寻最佳匹配的HMM,再用phmmer工具针对由最佳匹配HMM搜索每个query蛋白及对应的注释,见下面参考文献:
Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper | bioRxiv
相当于网站帮我们做了hmm search,我们只需要筛选最后一列,输入关键词WRKY,删除H列描述(Description)中可能与WRKY无关的基因,可以看到有50个候选的WRKY基因家族成员。这个时候就可以把A列(query)基因名提取出来,查找基因名对应的蛋白序列,复制整理出候选基因家族蛋白序列。

2. 通过hmm search鉴定基因家族
上面的注释结果中不一定有目的基因家族成员,这个时候需要我们直接从Pfam网站下载对应基因家族的HMM模型,或者使用多序列比对构建HMM模型,再进行基于蛋白结构域的比对寻找候选的基因家族成员。
2.1 使用TBtools的Simple HMM Search功能
TBtools这个软件功能还是比较全的,确实可以实现无代码做基因家族分析。但是我觉得还是有必要了解一下原理,毕竟软件是别人做的,过度依赖的话会导致自己处于被动的地位。
首先下载pfam全库的HMM模型:
https://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz
解压之后大概1.5个G的大小,这里面包含了所有HMM模型!
打开TBtools的Simple HMM Search功能页面:
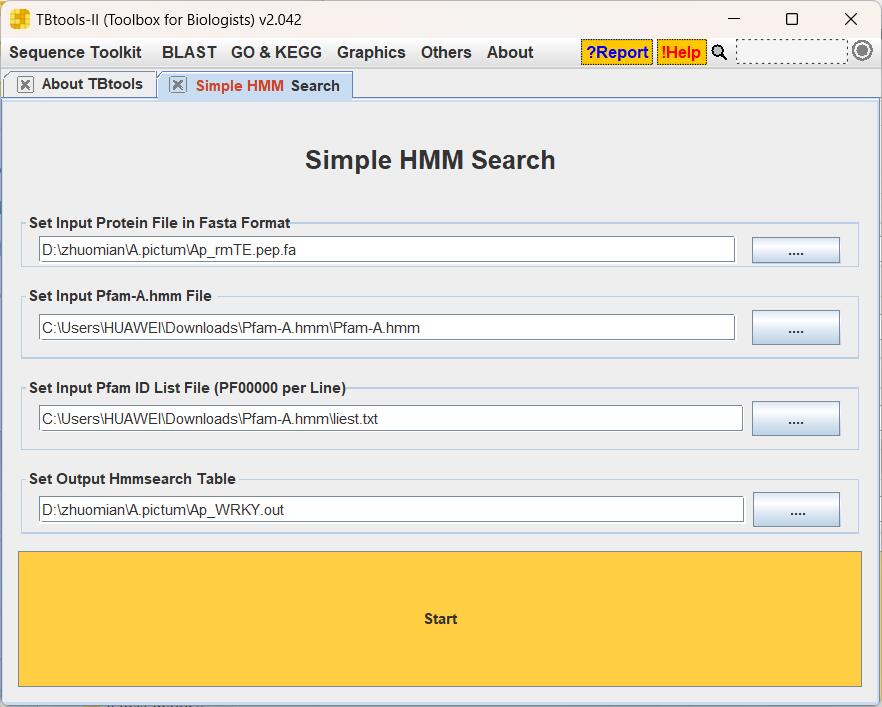
第一个输入框是物种蛋白序列文件。
第二个输入框是解压之后的pfam全库模型。
第三个输入框是你需要查找的HMM模型编号,比如我这里想要查找编号为PF03106的WRKY基因家族模型,就创建一个文件第一行写入PF03106即可(可以多个模型,一个模型编号一行)。一般通过文献中给出的HMM模型编号(PF号),或者在Pfam数据库直接搜基因家族对应的结构域信息得到模型编号。Search - InterPro (ebi.ac.uk)
第四个输入框指定输出文件位置和名称。
结果文件可以分为Sequence scores和Domain scores两部分,根据自己需要对E-value进行过滤。
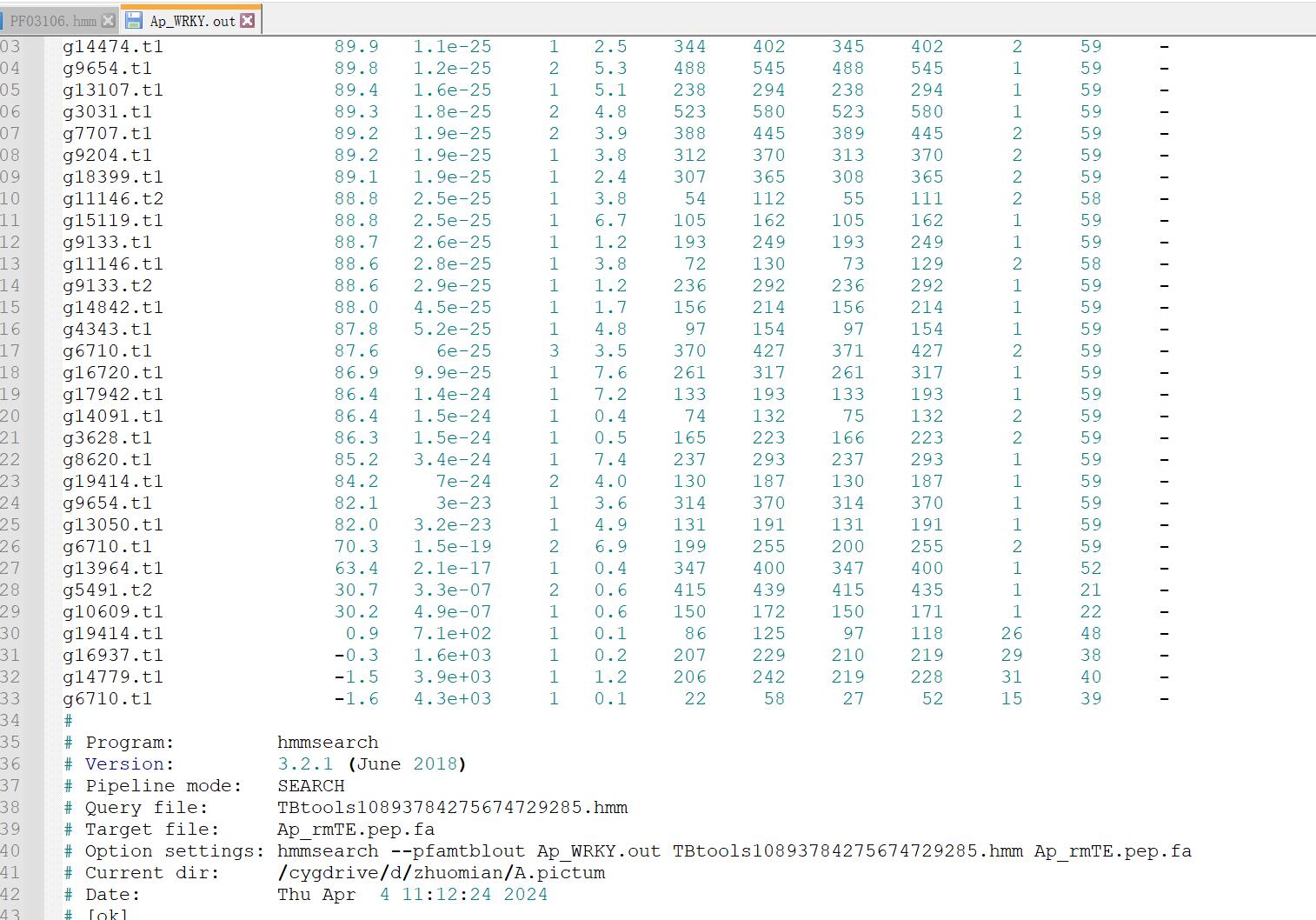
也可以看到这个工具就是调用了hmmsearch进行的同源序列搜寻。
如果很不巧你的基因家族在pfam网站没有相关的保守结构域,那就需要进行多序列比对构建自己的HMM模型了。
我在tbtools作者的知乎账号里有看到有使用多序列比对结果进行HMMsearch,但是在最新的2.081版本里没有看到相关功能插件…
插件 | 地表最强 Hmmer Search 界面工具 - 知乎 (zhihu.com)
不过不影响,我们可以直接用HMMER软件包。
2.2 使用HMMER软件包
使用HMMER软件包需要对以下两个主要程序有个大致的理解:
hmmbuild: 使用多序列比对构建HMM模型hmmsearch:使用HMM模型搜索蛋白序列库
还是以WRKY基因家族为例,在pfam网站上直接下载WRKY结构域的HMM模型(搜索PF03106,点击Curation、Download)

1 | hmmsearch --cut_tc --domtblout Ap-WRKY.out PF03106.hmm Ap_rmTE.pep.fa |
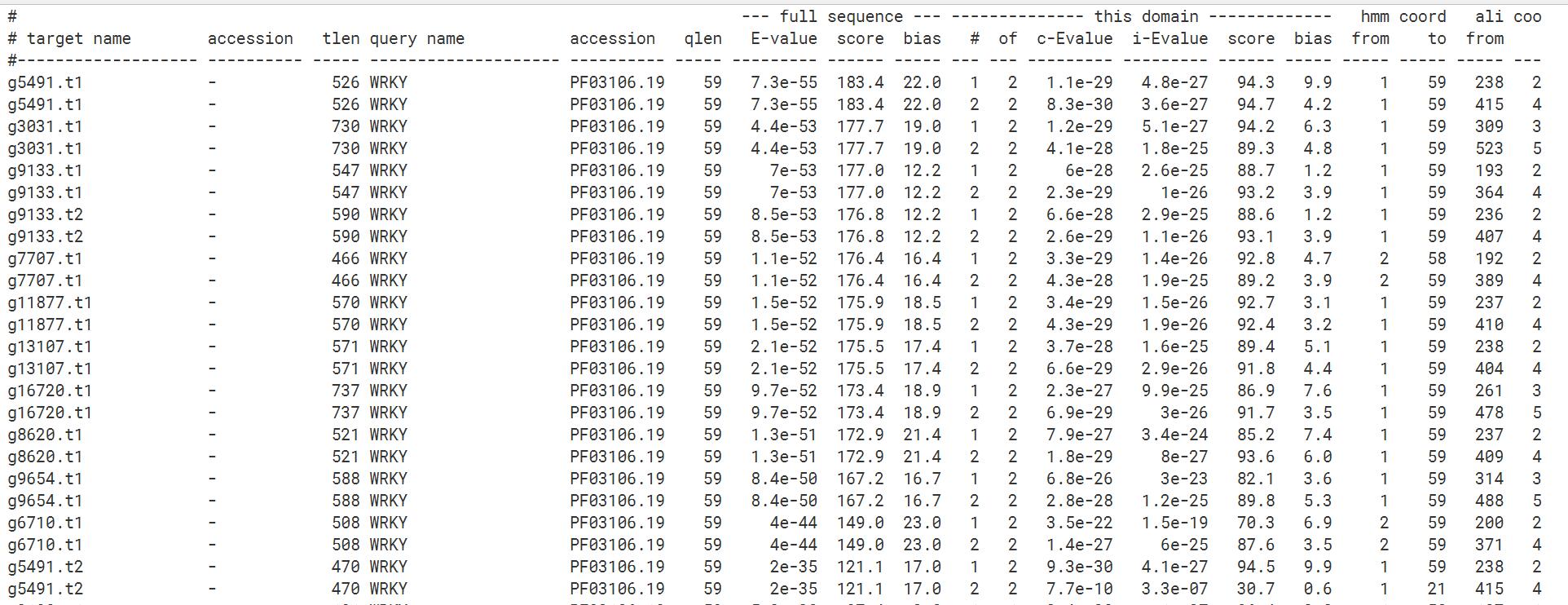
我这里用到的输出格式--domtblout,选择最后一个输出格式就和上面tbtools出来的结果完全一样了。
这里筛选出候选的基因家族成员后,我们还可以进一步的构建自己这个物种的WRKY基因家族HMM模型。对于前面说的,如果pfam网站没有你想要的保守结构域信息,我们也可以找到其他物种的同源蛋白,基于多序列比对结果构建HMM模型。
1 | grep -v "#" Ap-WRKY.out|awk '($7 + 0) < 1E-20'|cut -f1 -d " "|sort -u > Ap-WRKY-geneid.txt |

这一步构建的模型和你从pfam官网下载的HMM模型格式是一样的(这里除了GA/TC/NC参数,下面说),只不过用了我们自己的序列比对结果做为种子,训练多序列比对结果构建HMM模型。接下来使用构建的HMM模型扫描物种的蛋白数据库,进一步寻找候选的基因家族成员。
1 | hmmsearch --domtblout WRKY.out Ap_WRKY.hmm Ap_rmTE.pep.fa |
需要注意一点,如果你用的fa格式的多序列比对结果构建的HMM,是不包含GA、TC和NC参数的。只有用官方推荐的SELEX或者Stockholm格式(PFAM友好的多序列比对格式)才会有上面三个参数,否侧你用--cut_tc这样指定算法是会报错的!
一个可以转换多序列比对格式的网站,需要可以自取:
https://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_phylip.php
这种方式找到的候选基因家族数量比上面直接hmmsearch得到的数量多,也是需要自己确定阈值筛选的。
2.3 HMMER补充说明
HMMER软件包功能远不止上面的构建HMM模型和搜寻同源序列,顺便就再介绍下其他功能和操作。
一个蛋白可能有多种结构域,比如说我想确定上面候选的WRKY基因家族成员中还包含了哪些其他结构域该怎么办?这个时候可以用到hmmscan命令,使用蛋白序列搜寻HMM库。
1 | gzip -d Pfam-A.hmm.gz |

可以看到前面鉴定的WRKY基因家族成员中还有Plant_zn_clust这个结构域,这也是常见于WRKY基因家族中的一种锌指结构域。
顺便再提一嘴,HMMER也有线上的服务,见phmmer search | HMMER (ebi.ac.uk)
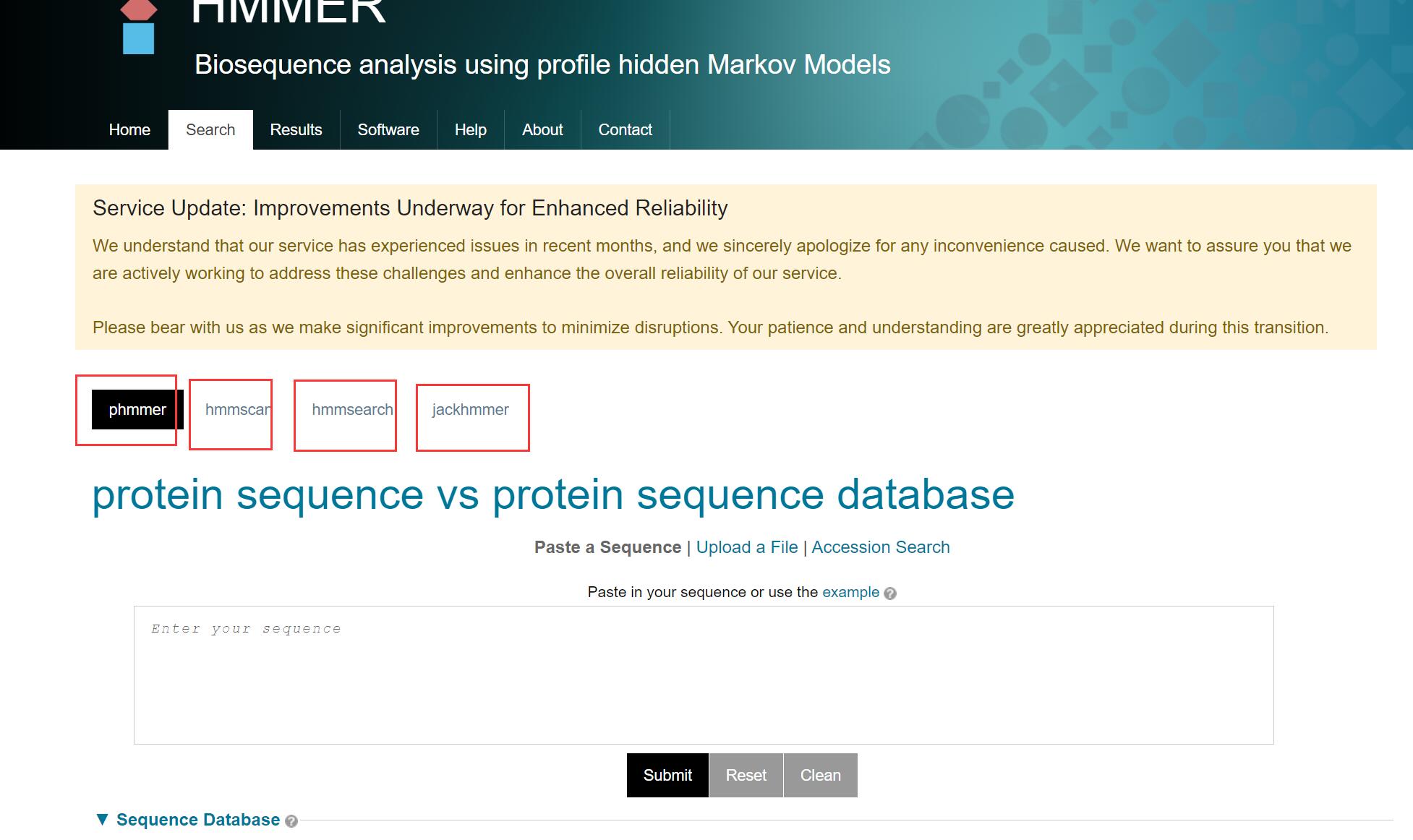
提供了包括前面说的hmmscan和hmmsearch两个功能,不过这两个功能不能指定你自己的蛋白数据集,只能用网上的数据库,比如Reference Proteomes、SwissProt、Ensembl这些。换句话说,如果你的物种比较小众,或者没有注释地比较完整的蛋白库,那你就不能通过这个网站去鉴定基因家族,也没法自建HMM模型。
如果你做的是模式植物,那么恭喜你,选择第一个高置信度的参考蛋白组数据库Reference Proteomes,指定特定的物种名即可。
再顺便说一下另外两个功能:
phmmer:与blastp类似,使用一个蛋白质序列搜索蛋白质序列库jackhmmer:与psiBlast类似,蛋白质序列迭代搜索蛋白质序列库
Jackhmmer是一种基于HMM的迭代搜索算法,可以使用一条或多条蛋白质序列,在蛋白质序列数据库中寻找同源序列。首先用输入序列构建一个初始的HMM模型,在指定的数据库中搜索,将这些匹配的序列,加入到输入序列中(也就是迭代的过程),重新构建一个HMM,并重复搜索过程,直到达到最大迭代次数或没有新的匹配序列为止。优点是可以发现较远的同源序列,提高敏感性和准确性。
官方都有示例数据可以在线跑,操作一遍就能理解了。
3. 验证基因家族
我们往往会用多种方法验证基因家族鉴定的结果,常见的是使用blastp。关于怎么使用blastp我就不多说了,可以见我以前的博客blastn & blastp 寻找同源基因
还有在线网站,比如NCBI的CDD数据库Home - Conserved Domains - NCBI (nih.gov)
可以使用Batch CD-Search功能模块,对鉴定的基因家族成员蛋白序列在进行一次结构域预测,结果如下:
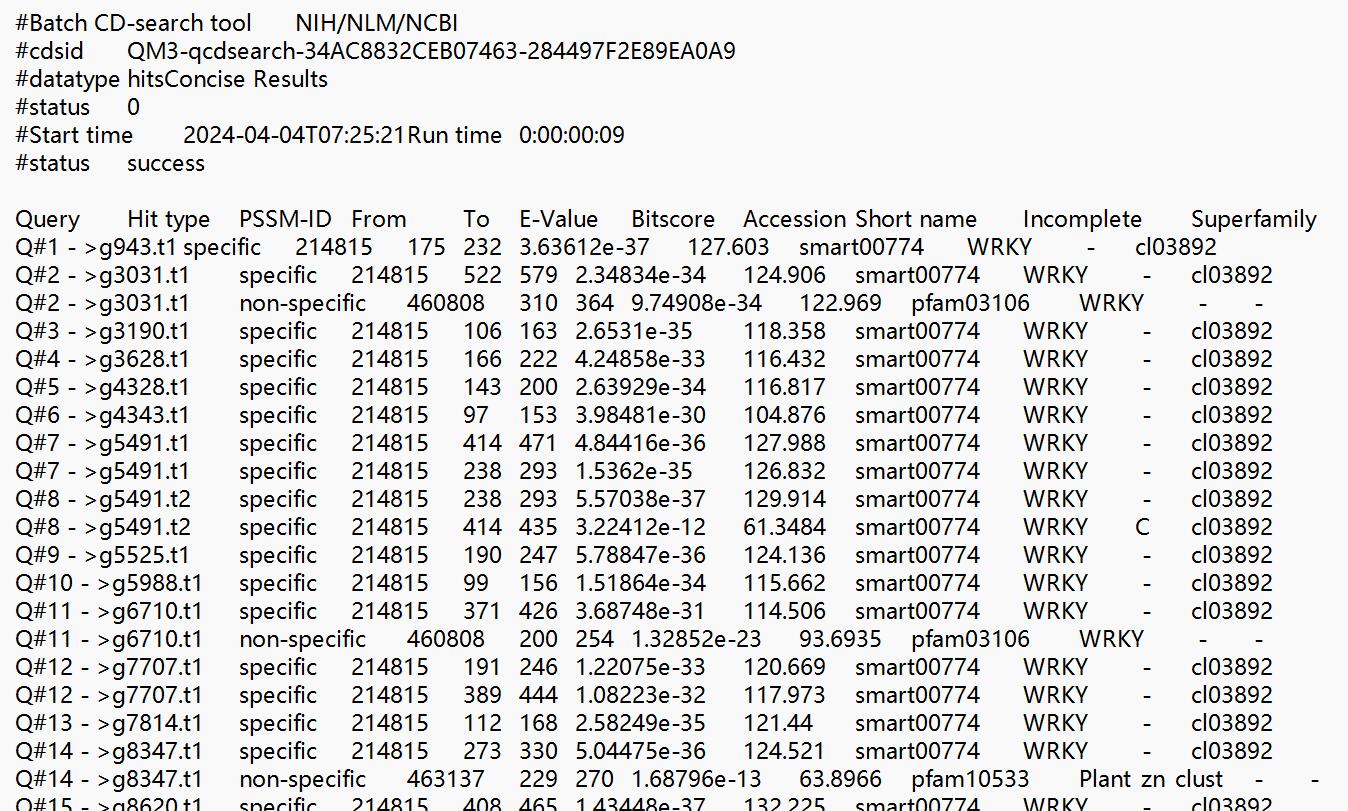
一方面可以验证鉴定的基因家族准确性,另一方面还可以用CDD预测的结果画保守结构域图,这个下次再说。