



最近用自己组装的植物基因组在做基因家族分析,简单记录下自己对数据的处理以及分析的流程。
随着现在测序技术的普及,越来越多的植物做了全基因组测序,对于基因组比较小的植物,现在纯做基因组组装和注释已经很难发文章了,一般来说我们还要再提出和解决一些生物学问题,最基础的就是通过比较基因组学,对系统发育中的代表性物种之间的基因家族进行比较分析、构建系统发育图谱,来揭示这些基因家族的起源和功能。
这篇笔记的流程主要是记录下使用OrthoFinder + r8s + cafe进行基因家族聚类、构建系统发生树、估算分歧时间和基因家族的收缩扩张分析。
数据准备
最重要的就是要拿到你想要分析物种的基因组数canX据,做基因家族分析的话蛋白质序列文件、CDS序列文件这两个是必需的。如果要做共线性分析(如MCScan’X),还要有gff文件,以及对组装的基因组有要求,一定是要组装到染色体级别(只有contigs的共线性分析是没有意义的)。
选择物种
植物基因组数据可以通过NCBI的genebank数据库、Ensemble数据库或者Phytozome数据库获得,选择的物种要和自己研究的物种亲缘关系不能太远,而且要在进化上有层级结构,染色体倍性最好一样,性状要能够说明一定的生物学问题。还要选择一个合适的外群,做进化树的时候要通过外群确定树根。
我们可以将已发表的近缘物种基因组文章中进化树作为参考选择需要的物种,或者结合网站Published Plant Genomes (plabipd.de)选择合适的物种。上面的网站至今仍然在更新,你可以很方便的找到已发表的植物基因组文章,得到这些已测序的植物在进化关系中的位置,如下所示:

我是一边参考这个网站,一边打开genebank数据库找到需要的植物基因组,下载蛋白序列文件和CDS文件(genbank有些植物基因组中没有相应的数据,需要从gbff文件中提取)。可以根据网站上显示的进化关系,提前写一个思维导图预计做出来物种树长什么样子(可以和后续做出来的进化树对比)。
数据预处理
预处理主要做提取最长转录本,以保证基因家族聚类的准确性和提高计算效率。在genebank中下载的几个物种蛋白质序列都是去冗余的,但是我自己组装的物种没有去冗余,可以用下面的脚本将isoform蛋白序列过滤,只保留最长转录本。
1 | # 蛋白序列提取最长转录本(开头有空行会报错!需要检查输入文件) |
一个简单判断自己蛋白序列是否冗余的方法:
1 | grep "\.2" 蛋白序列文件 |
如果基因名中有xxx.2的格式,说明这个蛋白序列不是去冗余的(正常情况下)。蛋白序列去冗余之后记得也要把相应的最长转录本的cds序列提取出来,写一个python脚本可以提取出来,这里就不赘述了(本篇笔记cds序列暂时用不到,后面做ks计算再说)。
1. 基因家族聚类&系统进化
基因家族聚类是将来源于同一个祖先,由一个基因通过复制而产生的一组基因进行分类和注释,对本物种特有的基因家族进行GO和KEGG富集分析(这个后续再说)。通过物种共有的基因家族中单拷贝同源基因构建系统发育树,用韦恩图可视化待分析物种间特有的基因家族和共有的基因家族数量。
把这两个放在一起的原因是OrthoFinder可以一步做到位。
OrthoFinder
davidemms/OrthoFinder: Phylogenetic orthology inference for comparative genomics (github.com)
按照github官方仓库中说的conda安装方法,运行的时候会报下面的错物:
1 | For Linux 64, Open MPI is built with CUDA awareness but this support is disabled by default. |
不知道为什么,OrthoFinder运行是不依赖于Open MPI的,总之无法正常运行,所以我下载了官方的source源码,解压后conda安装了必需的两个软件:
1 | 多序列比对软件mafft |
我们这里只需要将比较的物种蛋白序列放在一个文件夹中(比如mydata)就可以了,每个物种一个文件,以.fa .faa .fasta .fas .pep后缀都可以识别,前缀改成物种名。写一个slurm脚本:
1 | !/bin/bash |
-M msa指定物种树的推断方法,将使用串联的多序列比对进行最大似然树推断。该模式下会使用MAFFT进行序列比对,用FastTree进行树推断。
构建物种树有两种方法:串联法和合并法,orthofinder使用的是串联法,将所有单拷贝直系同源基因序列连起来,再进行多序列比对建树,该比对文件位于MultipleSequenceAlignments/SpeciesTreeAlignment.fa。合并法是将每个单拷贝基因家族建树,然后对基因树进行整合得到一致树(了解一下就行)。
结果文件
运行结束后可以在蛋白序列文件中找到OrthoFinder结果文件夹,结果文件非常多,官方有解释每一个文件夹信息:Exploring OrthoFinder’s results | OrthoFinder Tutorials
比较重要的几个结果文件:
MultipleSequenceAlignments/SpeciesTreeAlignment.fa 串联的多序列比对文件
Species_Tree/SpeciesTree_rooted.txt 含有bootstrap值的物种树文件
Orthogroups/Orthogroups.GeneCount.tsv 直系同源基因数目文件
Single_Copy_Orthologue_Sequences 单拷贝直系同源基因文件夹
Comparative_Genomics_Statistics/Statistics_Overall.tsv 统计文件
打开Statistics_Overall.tsv文件可以获得如下信息:
1 | Number of species 12 |
一共有468058个基因(占比93.1%)被聚类到39603个正交群(orthogroup)中,至少80%以上的基因可以聚类到正交群,说明这个结果还是很可信的。
打开SpeciesTree_rooted.txt文件可以获得进化树信息:
1 | (O.s:0.288854,(((L.s:0.323215,(N.a:0.233945,(C.c:0.185824,((A.p:0.00372873,A.v:0.00344612)1:0.102058,C.r:0.121885)1:0.0883246)1:0.0751893)1:0.0565903)1:0.0505776,(((G.h:0.0112911,G.b:0.011717)1:0.209852,A.t:0.363066)1:0.0376571,M.t:0.325608)1:0.0450521)1:0.0678736,A.c:0.272559)1:0.288854); |
去掉冒号前的1,就是标准的进化树文件类型.nwk,1是Bootstrap值(自展值),大于70%表示构建的进化树非常可靠,后续处理的时候需要把1手动删除。关于进化树的基础概念,可以参考:R语言绘制进化树:treeio+ggtree - 知乎 (zhihu.com)
我们可以用可视化树的软件,比如导入Dendroscope:
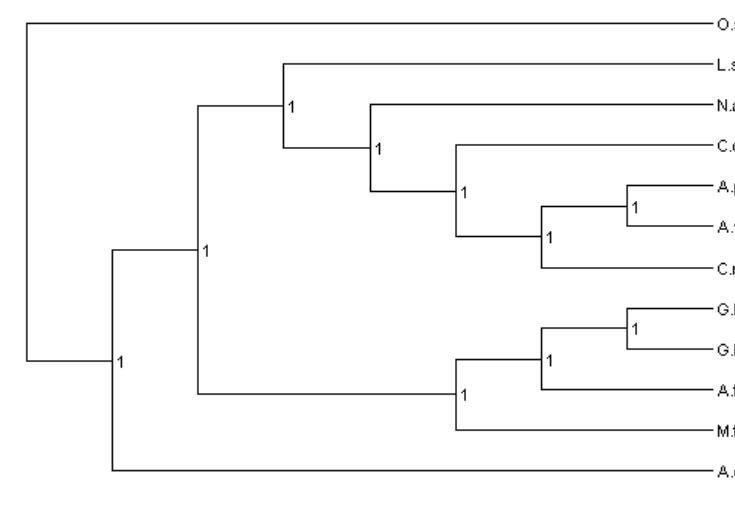
这个时候可以对比一下前面思维导图做的进化树,结构是一模一样的。
打开Orthogroups.GeneCount.tsv文件,可以看到各个物种在正交群中的基因数,这个文件后面做基因家族收缩扩张要用到。我们可以先简单整理一下需要分析的物种数据:
1 | # 删除第一行,然后选择第二列大于零的行,并将这些行的第一列保存到名为 1.txt 的文件中 |
awk '$2 >0 {print $1}'根据需要改哪一列,最终是一个物种一个文件,每个文件统计基因数量大于0的正交群,可以用在线工具可视化聚类结果。以在线VENN图为例:jvenn (inrae.fr)

2. 分歧时间估算
根据Species_Tree/SpeciesTree_rooted.txt文件中的进化树信息,我们借助化石时间矫正,可以得到有分化时间的物种树(超度量树/时钟树)。进行分歧时间估算的软件有PAML中的mcmctree命令,软件beast、r8s、Multidivtime等。
在算法上,mcmctree、Multidivtime和beast三个软件都是基于贝叶斯(Bayes)方法,mcmctree可以选择相关速率模型(correlated rate model)或者独立速率模型(independent rate model),支持蛋白序列或者核酸序列输入。r8s可以选择NPRS(Nonparametric rate smoothing)方法,惩分似然法(penalized likelihood,PL)等。
看到一篇文献比较了使用不同软件和模型构造时钟树,mcmctree在较浅的分歧估算中表现好,Multidivtime和r8s在较深的分歧估算中表现更好,而beast表现都不怎么好。参考以下原文:
(PDF) Dating the origin of the major lineages of Branchiopoda (researchgate.net)
考虑到r8s运算速度更快,以下用r8s进行分歧时间估算,有空再把mcmctree也跑一遍。
r8s
源码地址:r8s download | SourceForge.net
我下载r8s源码编译的时候会报错,后来在Biostars找到一个解决方法:
1 | tar -xzvf r8s1.81.tar.gz |
源码包中有使用手册,国内有翻译成中文的版本:r8s使用指南.pdf (book118.com)
使用r8s需要根据手册写一个批处理文件,你可以自己根据需要写,也可以用cafe5官方提供的prep_r8s.py
这个python脚本需要几个输入参数:
- -i 输入文件,也就是上面生成的树文件
- -o 输出文件,自定义的输出文件名称
- -s 对齐的序列氨基酸数量
- -p 校准的物种名
- -c 校准时间
注意!如果你是python2可以直接运行,如果是python3以上需要修改源代码的59行:
1 | print "\nRunning cafetutorial_clade_and_size_filter.py as a standalone...\n" |
python3之后print “”改成了print()。
1 | $ seqkit stat MultipleSequenceAlignments/SpeciesTreeAlignment.fa |
可以用seqkit工具(需要conda下载)计算对齐序列的氨基酸数量,这里是128458
校准时间可以通过物种分化时间查询网站TimeTree :: The Timescale of Life获得:
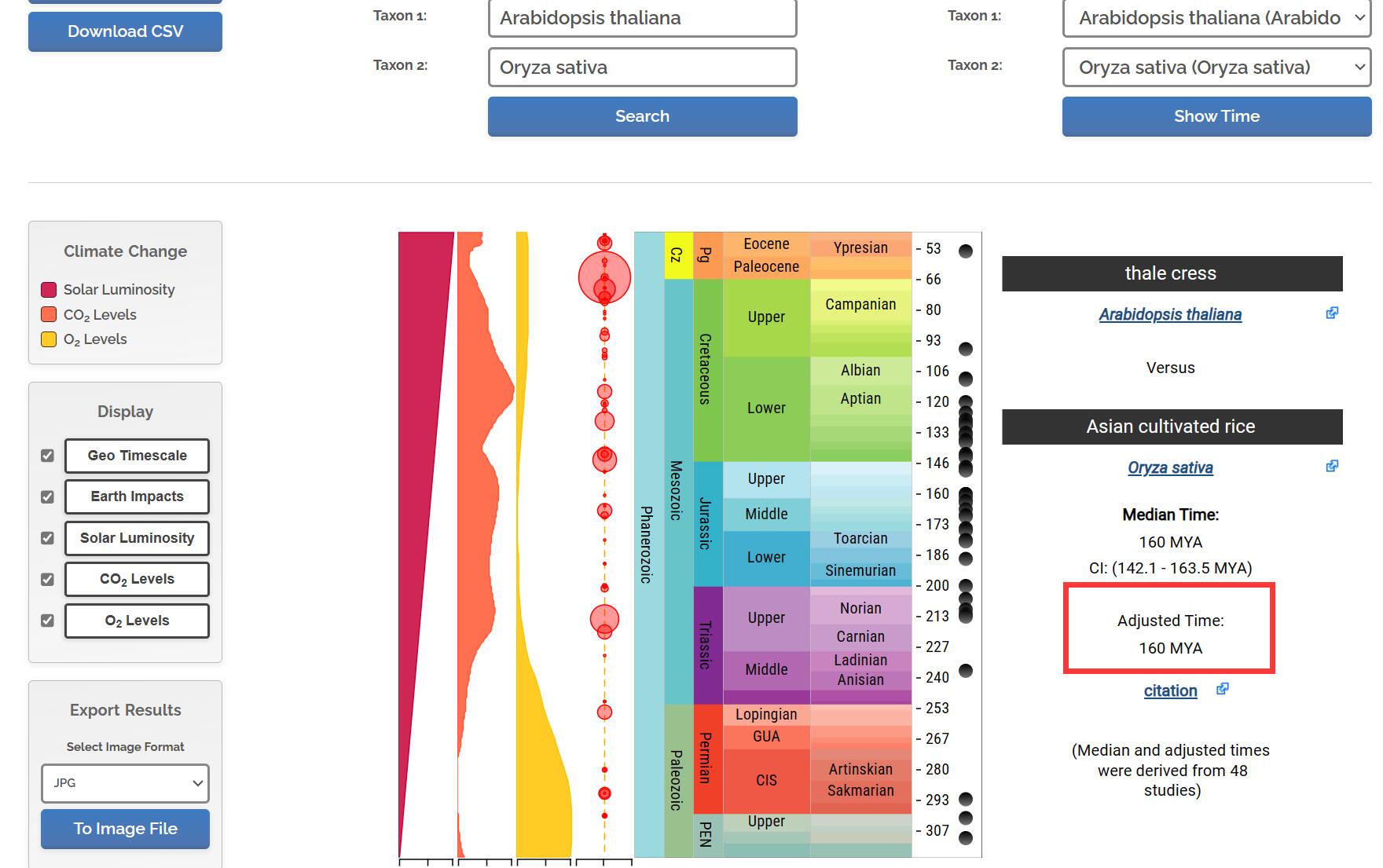
1 | python prep_r8s.py -i SpeciesTree_rooted.txt -o r8s_ctl_file.txt -s 125517 -p 'A.thaliana,O.sativa' -c '160' |
生成的文件r8s_ctl_file.txt就是r8s的批处理文件,其实这个python脚本就是指定了一些参数,可以看到定义了anaiva物种为假定的拟南芥和水稻的共同祖先(两个物种名的后三个字母组合),分化的时间为我们上面查的160MYA。
1 | #NEXUS |
有了批处理文件就可以运行r8s了:
1 | r8s -b -f r8s_ctl_file.txt >r8s_tmp.txt |
实际上我们只需要最后一行的时钟树,并手动删除假想的共同祖先anaiva(为了之后运行cafe):
1 | tail -n 1 r8s_tmp.txt | cut -c 16- > tree.txt |
这个tree文件可以在TVBOT (chiplot.online)进行可视化(网站上推荐是用mcmctree和beast结果),最后图片可以用AI做美化。
3. 基因家族收缩扩张
通过时钟树和基因家族的聚类结果,根据出生死亡模型估计每个分枝祖先基因家族成员个数,从而预测目标物种基因家族相对祖先的收缩扩张情况。
cafe5
hahnlab/CAFE5: Version 5 of the CAFE phylogenetics software (github.com)
cafe需要两个输入文件:
- 第一列为功能描述的直系同源基因数目文件
- 二元的,有根的,超度量树(nwk格式)
第二个文件就是上一步r8s生成的tree.txt树文件,第一个文件需要我们对Orthofinder结果文件中的Orthogroups.GeneCount.tsv做一些处理:
1 | 去掉最后一列total,添加第一列为(null),修改第一行Desc |
处理之后生成的cafe.input.tsv如下:

有两个物种我放的是四倍体,没拆成亚基因组,其他都是二倍体,可能会对单拷贝基因家族树统计产生影响。用官方仓库中的clade_and_size_filter.py脚本进行过滤:
1 | python cafetutorial_clade_and_size_filter.py -i cafe.input.tsv -o gene.families.filtered.tsv -s 2> filtered.log |
过滤超过100个拷贝的基因家族,得到gene.families.filtered.tsv文件后就可以运行cafe了:
1 | cafe5 -i gene.families.filtered.tsv -t tree.txt -c 10 -p -k 5 -o k5p |
cafe5和之前的版本不同,不用再写运行脚本,直接指定参数即可,-p指定根的频率分布为泊松分布,-k指定使用Gamma模型,这里指定了3种gamma rate,一般在2-5之间,-o指定输出文件夹名称。
如果你觉得自己做的物种进化速率不一致的话,还可以修改不同的lambda值,并指定参数-y:
1 | # 官方的例子,chimphuman_separate_lambda.txt,1和2分别代表不同进化速率的群体 |
结果文件
这里我指定了Gamma模型,结果文件结构如下:
├── Gamma_asr.tre # 每个基因家族树文件
├── Gamma_branch_probabilities.tab
├── Gamma_category_likelihoods.txt
├── Gamma_change.tab
├── Gamma_clade_results.txt # 每个节点的扩张/收缩基因家族数量
├── Gamma_count.tab
├── Gamma_family_likelihoods.txt
├── Gamma_family_results.txt
├── Gamma_report.cafe # 报告文件,用于下游统计分析、可视化作图等
└── Gamma_results.txt # Gamma模型的最终似然值,lambda值
cafe5刚更新的时候没有report.cafe这个文件,当时有人做了个可视化结果软件CafePlotter
moshi4/CafePlotter: A tool for plotting CAFE5 gene family expansion/contraction result (github.com)
这个软件近期仍然在更新,作图还是比较方便的:
1 | pip install cafeplotter |

这个图片的美化可以用Adobe illustrator,还有个可视化软件CAFE_fig:
我运行这个软件会报错module 'ete3' has no attribute 'TreeStyle',暂时无法解决。
## 2023/12/17更新
mcmctree
前面说到分歧时间估算除了用r8s之外,现在文章里最常用的就是mcmctree。正好这几天重新选了几个物种,顺便跑了一遍mcmctree,这里做一个记录。
因为mcmctree是PAML的一个子程序,所以直接源码编译安装PAML即可:
1 | # PAML的github镜像 |
这里编译的过程遇到一点小问题:
1 | cc -O3 -Wall -Wno-unused-result -Wmemset-elt-size -c baseml.c |
-Wmemset-elt-size这个参数只起到编译过程中忽略Warning信息的作用,我的gcc版本可能过低,不支持这个参数,在Makefile文件中直接把这个参数删除即可正常编译。
1 | # 将编译完成的二进制执行文件放到主目录的bin文件夹下,并把路径加入到.bashrc文件中 |
这样PAML就算安装结束了。
分析步骤前面是一模一样的,准备物种蛋白序列,取最长转录本,顺便筛选删除了50个AA长度以内的蛋白序列,跑一遍Orthofinder做基因家族聚类,记得加上参数-M msa,可以直接输出物种间单拷贝直系同源基因的比对结果(串联法)。
不是很明白,文献中好多是把Orthofinder鉴定的单拷贝直系同源基因又去跑一遍多序列比对,再去用别的软件建一个物种树,明明一个Orthofinder就可以包办这些事的。差别不大的话我个人觉得没必要去折腾……
mcmctree接收的是phylip格式的多序列比对文件,所以我们要对orthofinder跑出来的MultipleSequenceAlignments/SpeciesTreeAlignment.fa文件进行一点点修改(手动转成phylip)格式:
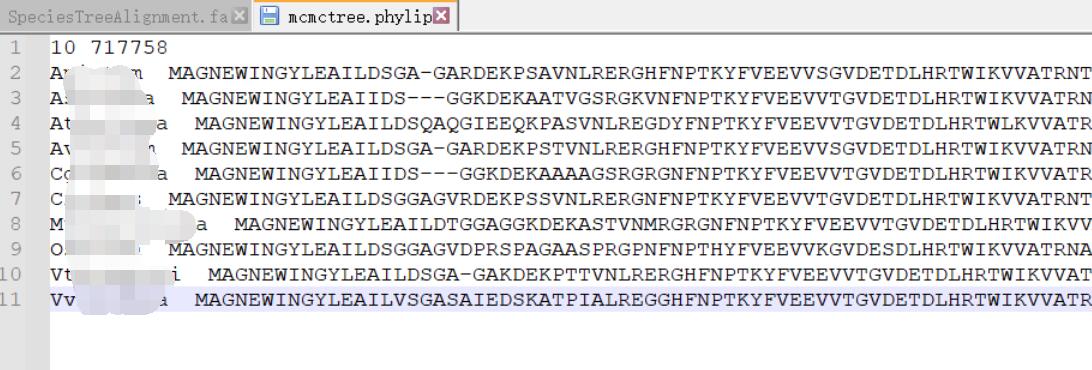
具体来说,第一行是物种数量 + 空格 + 比对的序列长度,后面每行是物种名(不能有.这个符号),两个以上的空格,以及比对的序列(一行的形式),简单写个python脚本处理一下:
1 | # 创建一个字典保存蛋白序列信息 |
和r8s一样,mcmctree估算分歧时间也需要化石证据做校准,区别在于mcmctree时间尺度是100个百万年(100Mya),比如我这里用了水稻和拟南芥以及拟南芥和葡萄的分歧时间做校准,也是用http://www.timetree.org/
mcmctree是在树文件中加入校准时间,并且要删除所有枝长、标签等内容,第一行加入物种数量,树的数量,直接拿orthofinder做的物种树Species_Tree/SpeciesTree_rooted.txt,修改后的物种树长这个样子:
1 | # 修改后的物种树命名为mcmctree.tree |
注意树中的物种名称和序列比对文件中的要一致,我这里隐去了物种信息,知道意思就行。
还需要修改一下配置文件,这里我参考生信技工的一篇文章,具体参数的意思就不重复造轮子了,估算系统树分歧时间 —— paml.mcmctree,r8s | 生信技工 (yanzhongsino.github.io)
配置文件如下:
1 | seed = -1 |
把dat/wag.dat这个氨基酸替换速率文件复制到当前目录下,跑第一次mcmctree mcmctree.ctl,生成out.BV这个文件。
1 | # 修改out.BV文件名为in.BV |
再跑一次mcmctree mcmctree.ctl,就生成了FigTree.tre这个分歧树的结果文件,这个文件可以直接在Tree Gallery (chiplot.online)可视化:
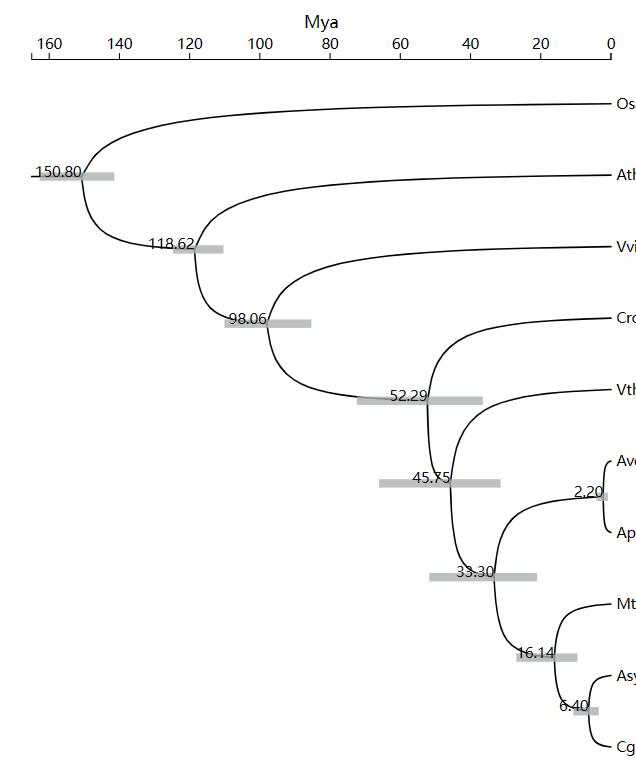
注意下时间尺度即可,还可以加上地质时间标尺等等,可以看开发者的b站教学视频:
小驰Coding的个人空间-小驰Coding个人主页-哔哩哔哩视频 (bilibili.com)
美化后加点AI调整,整理出图如下:
