最近在做非模式生物的GO和KEGG富集分析,参考了网上的一些帖子和知乎专栏,发现代码总有一些小问题,于是自己摸索修改终于跑通了= =,这里做个记录。
本篇笔记代码主要参考自:
比较基因组学分析3:特异节点基因家族富集分析(非模式物种GO/KEEG富集分析) - 简书 (jianshu.com)
小众或非模式生物的自建库GO/KEGG富集分析 - 知乎 (zhihu.com)
1. eggNOG注释
因为我们要做的物种是非模式生物,所有背景基因需要我们亲自注释,这里只推荐eggNOG,速度快,可以本地运行或者在线运行,在线运行有10万条序列的限制。在线网页的介绍可以看我的这篇笔记:基因组注释(6)——在线版eggNOG-mapper注释功能基因 - 我的小破站 (shelven.com)
假设我们已经拿蛋白序列做了注释,在一大堆结果文件中我们只需要out.emapper.annotation。用notepad++打开,去掉带“#”符号的前三行,把表头的query前面的“#”注释也给去掉,这些处理做好后文件如下:
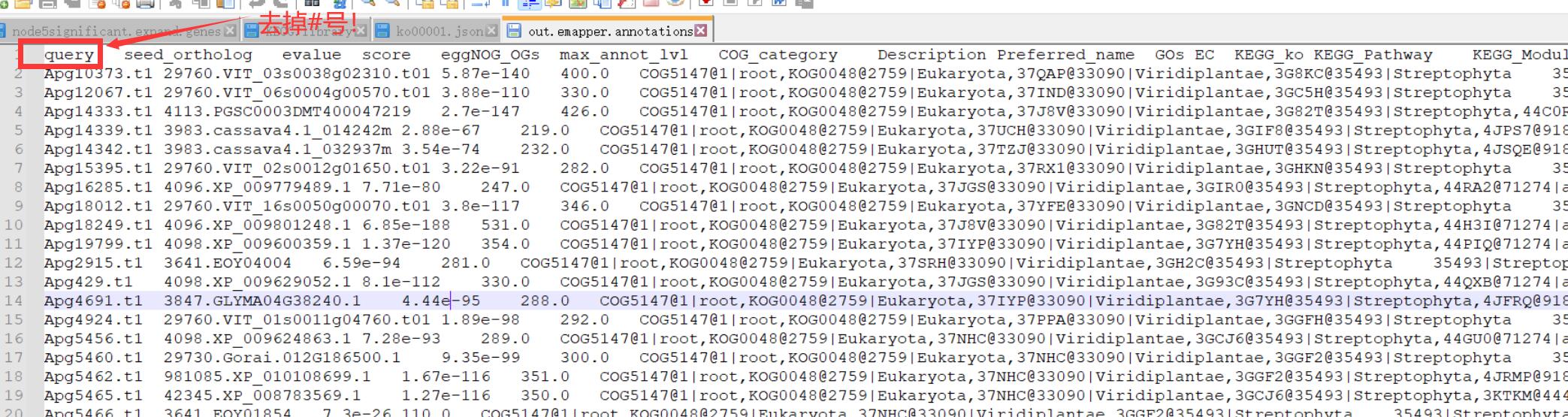
后续都是从这个文件中找到基因名与GO和KEGG号之间的对映关系。
假设我们从基因家族收缩/扩张分析、转录组等等数据已经拿到了我们想要的序列,创建一个新文件,一行一个基因名放入我们要分析的序列,文件命名为gene.txt如下所示:

2. GO富集
首先进行GO注释库的构建:
1 | # 下载最新的GO库注释 |
构建的GO注释库如下:

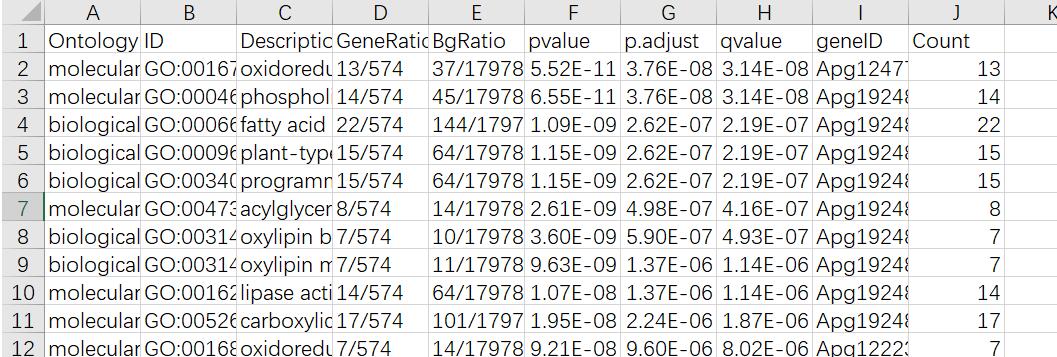
第一列是GO号,第二列是描述(注意要完整的描述,而非一个单词,否则后续因为描述重复而无法富集!),第三列是GO分类。
1 | library(clusterProfiler) |
看一下生成的GO_enrichment.xls文件如下:
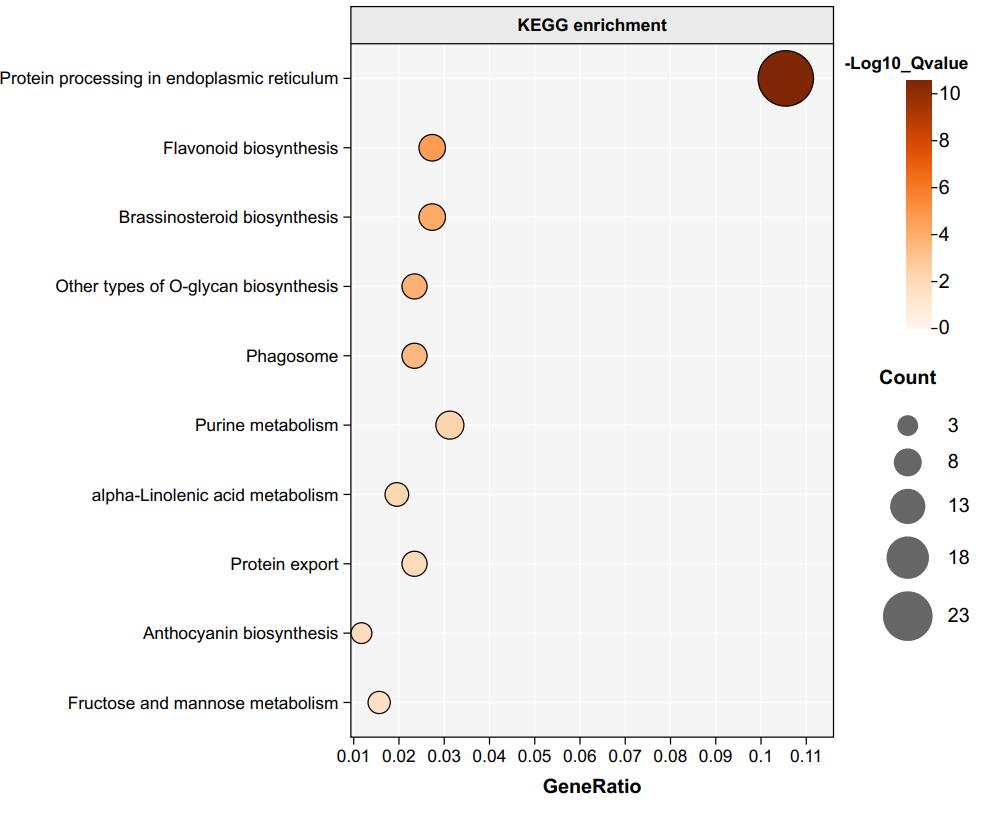
我们一般拿到这个文件后,根据qvalue值从小到大筛选可信度最高的富集结果,再手动去掉不合理的(比如你做植物,却注释到动物和微生物的)。
做到这一步生成GO富集文件就可以了(再做就不礼貌了),GO富集结果可以在R做可视化,但是我觉得没必要,真的,调参数真的太难受了,而且每个人作图的要求都不一样。我个人比较建议在一些在线网站上手动画,比如这个网站:ChiPlot
网站的开发者在B站传过可视化富集结果的教学视频:【ChiPlot】绘制KEGG/GO富集图_哔哩哔哩_bilibili
省流不看版
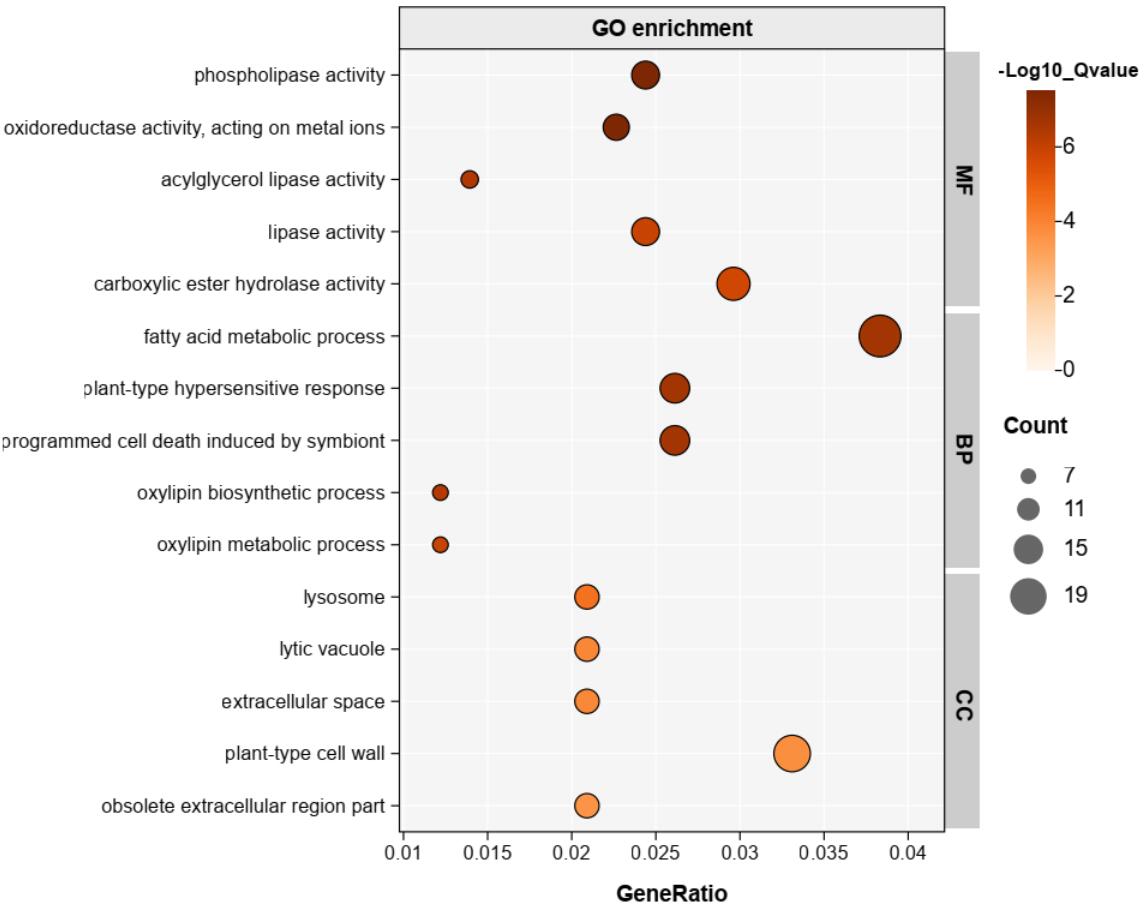
根据富集结果,筛选molecular function、biological process和cellular component三个类别中qvalue值前5的GO term,计算第四列的GeneRatio值。
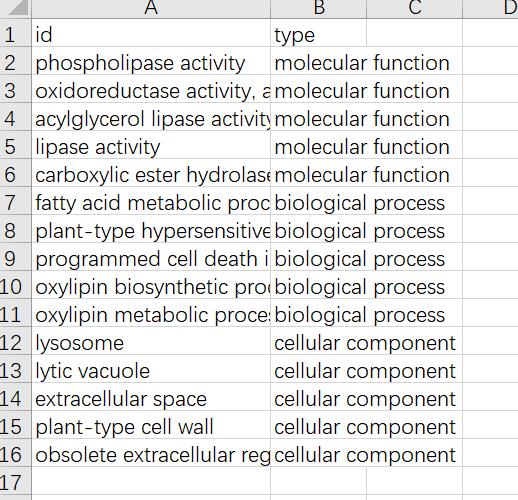
创建一个新的excel表,需要5列内容,如下:
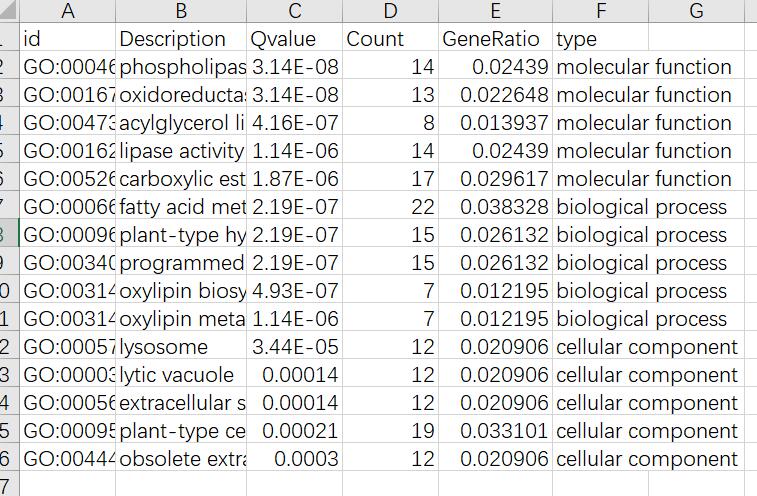
type那一列不需要,只是为了方便展示,id这一列的列名可以随意,Description、Qvalue、Count、GeneRatio四列列名必须要一样,顺序无所谓。
再做一个展示图层的excel,作用是区分GO term属于哪个类型:
传到上面网站的KEGG/GO富集作图的模块,手动调整参数(自己多试一下)再加点AI作图,反正比我R做的好一些:
3. KEGG富集
思路和GO注释是类似的,相比GO,KEGG可选择的物种会多一些,我们这里仍然当作是非模式生物,以第一步eggNOG注释的基因集为背景基因集。
构建KEGG注释库:
1 | # 下载KEGG注释库 https://www.genome.jp/kegg-bin/get_htext?ko00001,点击 Download json,下载得到ko00001.json文件 |
构建的KEGG注释库如下:

第一列是ko号,第二列是ko号对应的代谢通路描述,同样的,需要注意一个基因可能对应多个ko号,在对映关系上需要做一些处理:
1 | ## KEGG注释生成 |
最终得到的KEGG富集结果KEGG_enrichment.xls如下:
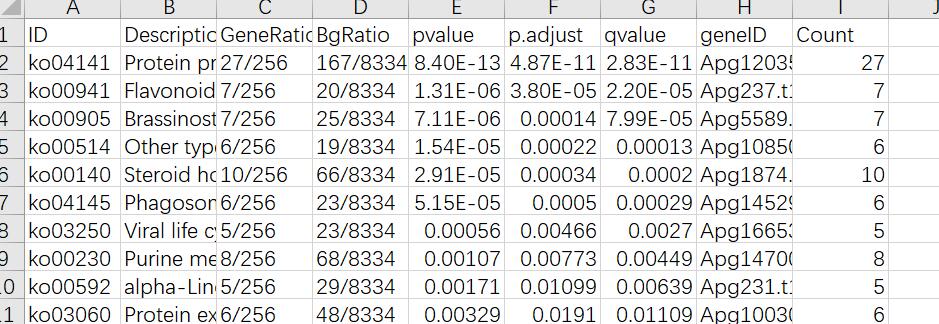
同样是根据qvalue筛选,剔除不合理的富集通路(比如你做的植物,富集到动物、微生物等代谢通路),作图是和GO富集作图一模一样的,这里不赘述了。
剔除不合理的代谢通路
顺便说一下,KEGG结果中会有不少不合理的地方,比如我注释的植物基因代谢通路,会有挺多注释到非植物相关的KEGG通路……
当然,我们可以一个个在KEGG官网查询富集的通路信息,也可以在R中把KEGG的所有植物代谢通路整理出来。这里需要用到一个R包KEGGREST快速获取KEGG数据库信息,需要注意,通过BiocManager下载的KEGGREST包已经不能正常使用了(2022年6月1日调用API地址发生了变化),需要在github上下载最新版:
1 | # 国内或许会出现网络问题,需要一点魔法(摊手) |
参考代码:玩转KEGG (二)——植物富集到了动物通路?难不成咱研究的植物人儿😂 - 知乎 (zhihu.com)
具体思路是用KEGGREST包抓取植物物种信息,获取每个KEGG库中植物的代谢通路,取并集:
1 | org <- data.frame(keggList("organism")) |
每一个物种都要抓取ko号,所以时间会有点慢,大概10分钟左右。
可以看到现在KEGG中一共有152个植物,取代谢通路的并集后,每行输出结果。
最后用我熟悉的python简单处理过滤一下:
1 | with open('plants.kegg.txt', 'r') as file: |
费这么老大劲功夫不如上KEGG官网把前几个代谢通路搜一下…..(想给自己一嘴巴子)
KEGG PATHWAY: ko03250 (genome.jp)
把网址上的ko改一改,看一下description就行…不过有的描述不是很清楚,这样过滤一遍准确性肯定是提高的。
最后把KEGG富集结果拿到Chiplot上做图即可: