



经历了三个月小论文+大论文的摧残,最近终于闲下来了一点,继续更新一下学习笔记~
今天主要记录下怎么做的转录组趋势(时序)分析。大多数时候,我们的转录组数据不仅仅只有一组处理组和对照组,比如梯度实验会设置不同处理浓度,或者同一浓度处理下设置不同取样时间,来观察取样组织中基因随着浓度、时间等的变化规律,也就是进行基因表达的趋势分析。
我们知道转录组测序分析的核心是获得每个样本基因的表达量,转录组趋势分析也是一样的。转录组分析的处理组和对照组中往往能鉴定成千上万个发生差异表达的基因,画成火山图会有密密麻麻的点,很难得到有效信息。趋势分析可以有效缩小我们的研究范围,将这些成千上万个差异基因按照表达趋势(比如同时上调,同时下调等等)进行聚类,同一种表达趋势的差异基因聚在一个cluster中,后续还可以再对感兴趣的cluster进行富集分析,进行功能注释等等。
现在的测序公司会把你送的材料做两两对照,每一个处理组和其他组之间都会丢给你一个差异基因表达量统计的excel表,这里面很多信息是用不到的= =,我感觉只是测序公司为了体现工作量而已。。。。。。跑题了,不管是送公司测转录组还是自己做转录组,我们都要明确一点,我们需要的分析的是差异基因(当然也可以是你感兴趣一部分基因),而不是一股脑儿把所有基因表达量都搬上去做趋势分析。
举个例子,我有个干旱胁迫的转录组测序数据,分别是胁迫发生的0h、4h和12h三组处理,我需要研究胁迫发生的这三个时间段叶片组织中差异基因有什么样的表达趋势。公司测转录组就很可能把所有组两两比对一遍,而我只需要 4 h vs 0 h 以及 12 h vs 0 h,这两组数据进行差异基因筛选,比如筛选标准是 |log2(foldchange)| > 1, FDR < 0.05,接着取两个组差异基因的并集进行后续趋势分析。当然,你自己用下机数据分析的话可以用Deseq2R包先去做个差异基因筛选,详细操作可以见我这篇博客,这里不罗嗦了。转录组数据分析笔记(7)——DESeq2差异分析 - 我的小破站 (shelven.com)
网上有很多在线平台可以做趋势分析,但是!我 都 不 推 荐!
原因很简单,这些平台都需要排队、收费等等,提高了一个很简单的分析流程门槛,而且这些平台都是基于STEM免费软件做的,吃相未免有点难看了。不如直接用官方软件。
1. STEM
Short Time-series Expression Miner (STEM),翻译过来叫短时间序列表达挖掘,原先开发的时候就是为了做基因表达时序分析的。官方建议的时间点(也就是处理组)不超过8个,因为超过8个组表达趋势会过于零碎和复杂,这种情况下可以做WGCNA分析,暂且不表。
安装STEM需要在windows系统上先安装JAVA。STEM官网如下:
STEM: Short Time-series Expression Miner (cmu.edu)
下载之后,双击stem.jar即可运行,下面是软件的UI:

参数虽然有很多,但是很多都是可以用默认的。
1.1 Expression data info
导入前面取的差异基因并集,比如说我用 |log2(foldchange)| > 1 的标准进行差异基因筛选,就可以整理以下tsv表格:

分别代表4 h和12 h两个时间点相对对照组的差异基因表达倍数变化趋势,如果你有重复组,还可以点Repeat Data按钮进行补充。
导入数据有三种标准化方式,如果你用的是表达量(比如TPM、FPKM),可以使用第一个log2标准化,或者第二个表达量差值的标准化。这两个标准化方式都是以第一个时间点为对照,一个是取表达量差值的log值,一个直接取差值。
如果你和我一样用的是表达量倍数(一般情况下做趋势分析也是关注表达量倍数的变化)就选第三种,也就是不进行标准化,因为我们用的已经是相对对照组的变化倍数了。第三种标准化方式会添加一个表达量为0的虚拟样本,实际上并不存在,可以理解为我这里的0 h对照组。
1.2 Gene info
这一栏主要是做每个模块(cluster)内基因富集注释用的,可以选择官方注释数据库,或者提供基因注释文件。我这个是非模式植物,后续需要手动构建KEGG和GO注释库进行富集分析,这里也暂时不表。
1.3 Options
这一栏是对聚类参数进行调整的,可以选择聚类方法,包括stem的聚类方法或者是K-means聚类方法。
主要的参数是Maximum Number of Model profiles,这个参数决定我们的差异基因最多可以分为几个cluster。不要用默认参数50!我这里相当于有3组,只需要设置8个cluster就足够了,数量太多反而会让趋势碎片,不方便我们进行趋势分析。
Maximum Unit Change in Model Profiles between Time Points这个参数是设置一个cluster模型可以在不同时间点改变的最大单位,改变这个参数会影响最终分组后的p值,这个问题不大,用默认即可。也可以看看用户手册有官网的详细描述。
在Advanced Options设置中可以设置基因的筛选条件、注释文件的细节等等,如果前面选择的聚类方法为K-means的话,也可以点击去调节细节,这里也不赘述了。
1.4 Execute
前面的参数设置完成之后,点击这一栏黄色高亮的Excute即可。
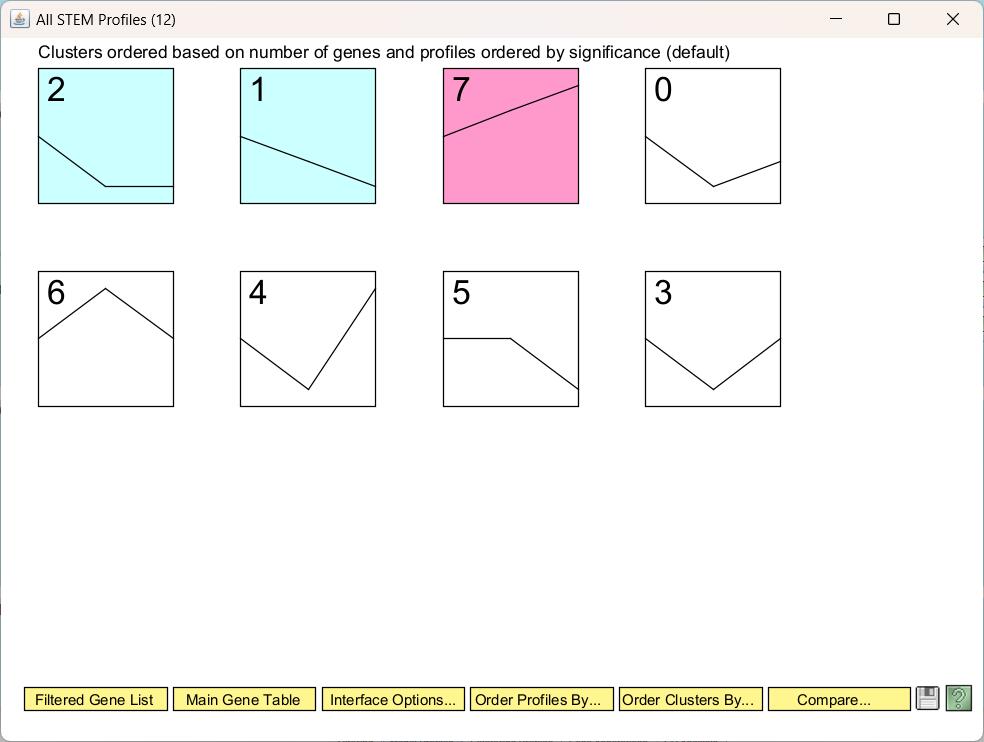
点击每个cluster可以看详细的信息,包括这一个cluster中的基因数量,每一个基因的表达模式,还可以导出每一个cluster的基因信息。
我们可以点击底下黄色高亮的Order Profiles By设置cluster的显示方式,比如可以加上p值(Significance,显著性特征),可以看到这些有颜色的cluster是p值比较低,也就是置信度比较高的。
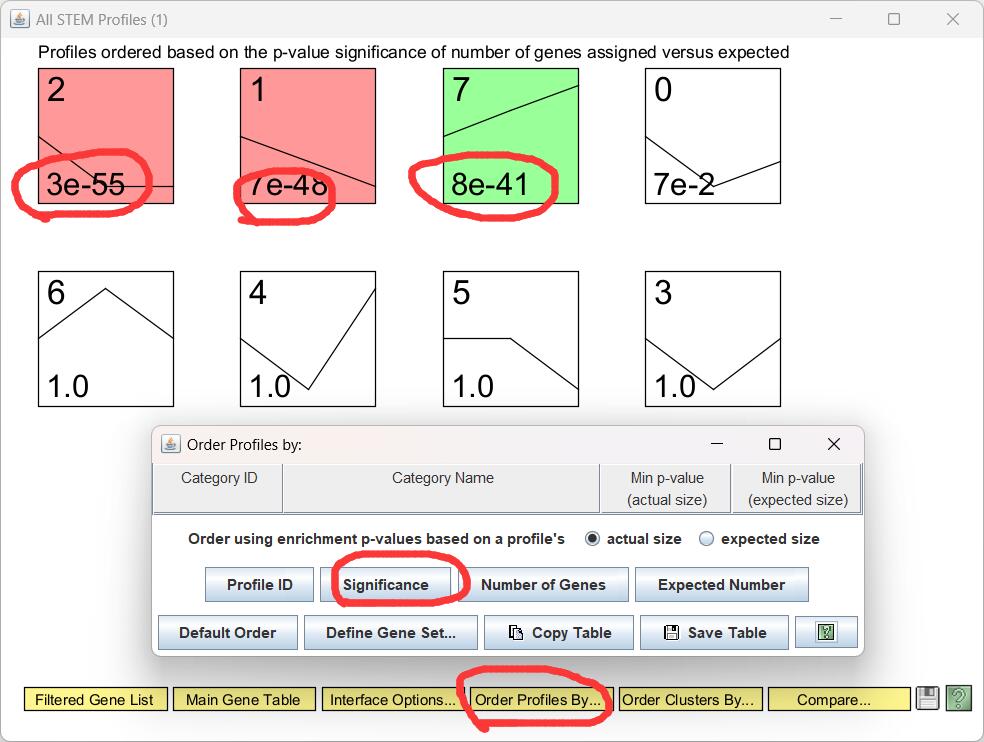
需要注意,那些p值为1的cluster不代表没有意义的表达趋势,还是要看我们感兴趣的差异基因在哪个模块,或者对我们感兴趣的基因对应地做分析!
2. ClusterGVis
上面这种转录组趋势分析图放在10年前可以说比较有亮点,因为STEM软件是2006年开发出来的,至今虽然也有大量的转录组分析文章在用,但是在今天看来,它能展示的信息量还是有点捉襟见肘。
介绍一个中国药科大学的博士师兄做的R包ClusterGVis:
junjunlab/ClusterGVis: One-step to Cluster and Visualize Gene Expression Matrix (github.com)
这个R包可以一步将趋势分析结果与基因的表达量结合一起,并且如果你自己有做每个cluster基因的富集分析的话,还可以把富集结果放在一张图中进行展示。作者在github上展示了这个R包的详细用法,我这里只做个人使用记录,向大佬表示感谢!!!
2.1 准备含有标记基因的基因表达量矩阵
比如我这里的TPM.marker.tsv文件,格式如下:
1 | 0h 4h 12h |
我这里用的是各个组的差异基因TPM表达量,对于自己感兴趣的基因,重命名为Ap开头的基因,也就是标记基因(比如差异基因里有你感兴趣的基因家族)。当然,如果你没有感兴趣的基因也可以不标记。这个表达量矩阵是做热图用的,也是做cluster聚类的基础。
2.2 cluster聚类
1 | setwd("D:\\zhuomian\\WRKY\\06.transcriptome\\clustergvis") |
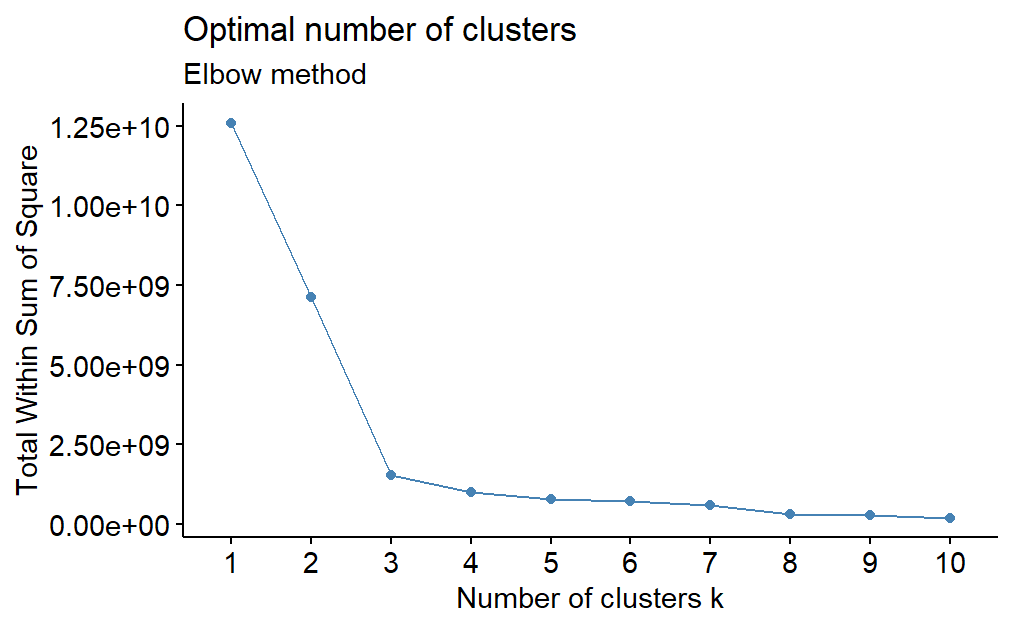
这里可以选择使用K-means或者mfuzz包进行聚类,getcluster函数出图如下,可以帮助你确认聚类个数,我这里选择8:
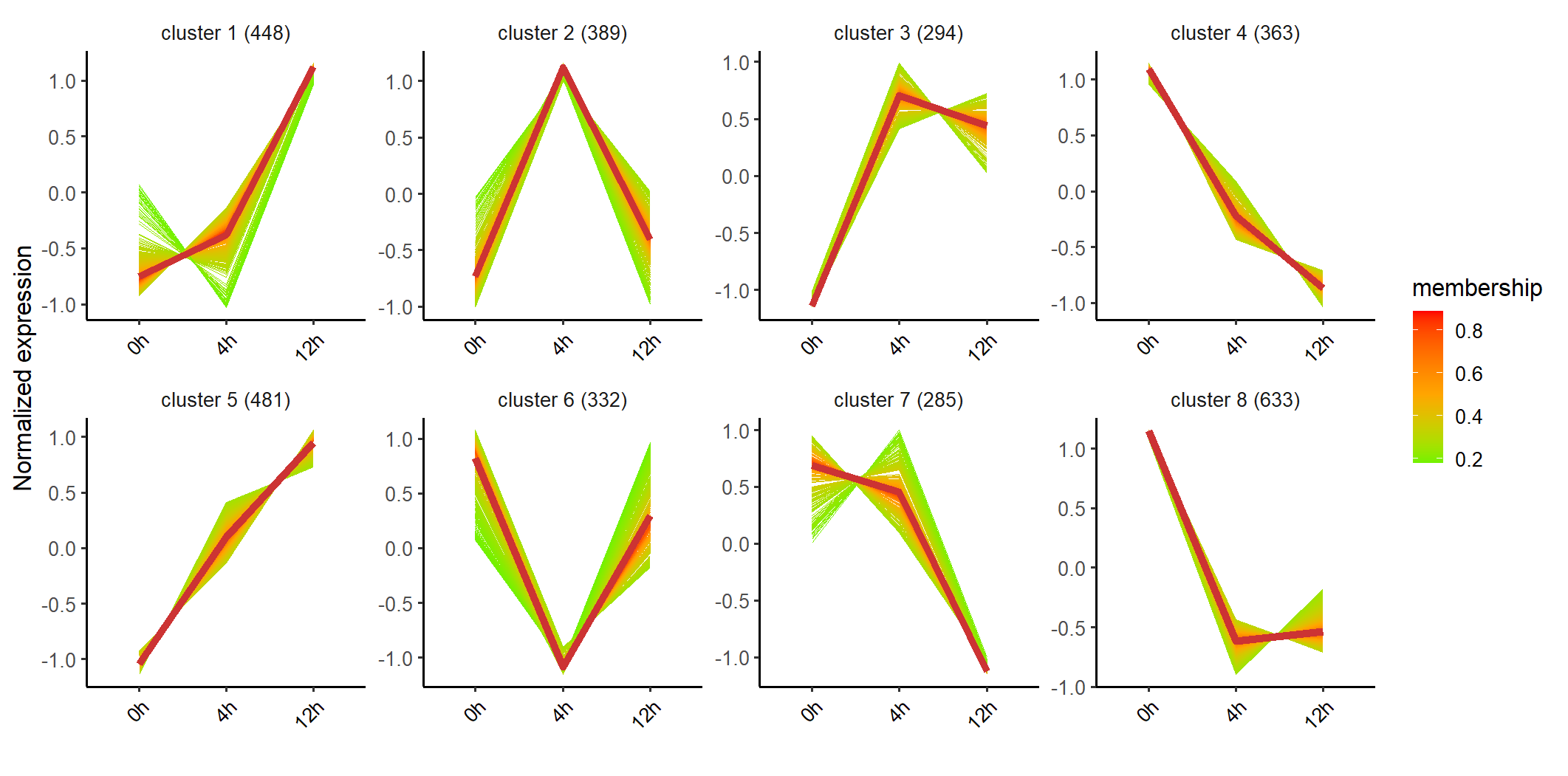
绘制的聚类折线图如下:
2.3 每个cluster进行富集分析
前面的R代码中,我们把聚类结果保存在了cm变量中,可以点开这个变量看具体的聚类结果:
简单来说,cluster聚类结果在cm[["wide.res"]][cluster]中,同一个cluster中的基因用同一种数字表示。
一个很笨但是有效的方法,把每个cluster的基因名调出来,每个cluster基因都做一遍GO和KEGG富集分析,非模式生物构建方法可见上一篇博客非模式生物的GO和KEGG富集 - 我的小破站 (shelven.com)
我们不需要展示所有富集结果,简单来说,可以将cluster富集结果分别做一个KEGG和GO统计结果表如下:
1 | # 文件cluster.all.GO.tsv(每组取前五富集结果) |
2.4 组合趋势聚类图、表达量热图以及注释结果
就是调参数的问题啦~
1 | # 添加标记基因(就是判断你要展示的基因是哪些行,这里我是Ap开头的基因) |
直接出图结果如下:

这样的出图基本上是不用改的,最多用AI调一调位置,大小。或者在R里调一下表达量热图的三个颜色:
ht.col.list=list(col_range = c(-2, 0, 2),col_color = c("#81C7C1", "white", "#FC7C5A"))
根据自己的数据调整一下就行,这种出图质量直接用于文章是完全没问题的,而且展示的信息量也足够多~