



整理一下前段时间做的基因家族分析笔记,这部分分析用的代码部分比较少,因为有现成的软件以及网站可以分析,没什么太多需要自己创造的地方,按部就班的分析流程实在让我提不起兴趣……简单记录下需要自己整理数据和写代码作图的部分——基因家族顺式作用元件预测。
关于怎么鉴定基因家族,以后有空再更新吧,以及基因家族成员蛋白保守结构域和基序的分析、基因家族在染色体上的分布、同源基因共线性关系等等这些都非常容易,这里就不赘述了。做到这一步,假设都拿到了基因家族成员对应基因组中基因的名字。
1. 顺式作用元件概念
还是要先了解一下概念:顺式作用元件(cis-regulatory element,CRE),是与结构基因串联的一段非编码DNA序列,起到调节邻近基因的转录作用(cis的拉丁语意思就是同一侧,即与调节的基因在同一段DNA序列上)。
顺式作用元件并不是只存在于转录位点的上游!最早的顺式作用模块的定义最初用于描述增强子,可以见下面这篇文献:
A systems biology approach to understanding cis-regulatory module function - ScienceDirect
如今顺式作用元件(或者称顺式作用模块,CRM)定义为具有转录因子结合位点的DNA序列,包括了启动子、增强子、沉默子、绝缘子等等,所以它可以在基因的上游、下游甚至基因内发挥作用!
而现在的基因家族顺式作用元件预测,清一色取基因上游1000 - 2000 bp左右序列,这或多或少有失偏颇。2000 bp的长度只是包括启动子区域(如果是细菌的话,2000 bp甚至可能取到其他基因区域了),而离转录起始位点比较远的,比如增强子就没有办法检测到了。所以我认为真核生物的这2000 bp长度,只能说是启动子区域的顺式作用元件预测。
2. 提取起始密码子上游序列
上面说了这么多,其实就想吐槽一下这样的公式化论文太多……做科研的同时要想想我们这么做的目的是什么,而不是一味套模板
回到正题,我们也取起始密码子上游的2000 bp序列,做基因家族启动子区域的顺式作用元件预测。
使用seqkit软件从fa格式的基因组文件和gtf格式的注释文件中提取起始密码子上游序列:
1 | seqkit subseq --gtf Ap_rmTE.gtf --feature start_codon --up-stream 2000 --only-flank --gtf-tag transcript_id /public/home/wlxie/Genome/Ap.fasta > upstream.fa |
得到的上游序列格式如图,基因名会在最后加上指定的tag名:

根据tag和自己需要分析的基因名之间的对应关系,可以把自己需要的基因的上游序列挑出来,顺便可以重命名一下。
3. 启动子区域顺式作用元件预测
将挑选的基因启动子区域序列(fasta格式)上传到PlantCARE进行顺式元件预测:
PlantCARE, a database of plant promoters and their cis-acting regulatory elements (ugent.be)
结束后会收到一封邮件,附件中有预测结果,我们只需要plantCARE_output_PlantCARE_5730.tab这种格式的汇总文件,这个文件可以重命名为xls格式,就可以在excel中打开编辑了。
我这里50个基因,预测的启动子区域顺式作用元件有8718个,需要根据自己实验进行数据过滤。举个例子,我想要知道这些启动子区域的顺式元件有哪些功能,我的筛选标准是:
- 删除CAAT-box和TATA-box,因为这两个是转录起始必须的,在功能上对我的分析内容没有任何帮助
- 删除第二列没有命名的顺式元件
- 删除最后一列没有功能注释的顺式元件
这样删除后8718个顺式元件就只剩下了1230个,平均下来一个基因有功能注释的启动子区顺式元件有20个左右,在可以接受的范围。
将最后一列功能注释找个翻译软件翻译一下,一共也就40个左右类型,根据功能描述,将它们进行分类以及最后一列批量替换,如下:

D列是起始位点,E列是顺式元件长度,F列是正负链,G是预测的物种,H是改后的功能名称。
简单来说,我将这些顺势元件的功能分为了光反应相关(light_responsive)、激素响应相关(hormone_responsive)、生长发育相关(development_related)和环境胁迫相关(environmental_stress)共四种类型。
3. 顺式作用元件统计图和堆积柱状图
看到有的分析会把顺式元件做个结构图显示出来,比如下面这样的(做两个bed文件,然后在GSDS网站中可视化):

我个人觉得实在意义不大,这种处理相当于是把这2000 bp序列看作是一个基因序列,每个顺式作用元件相当于外显子给可视化出来。且不说顺式元件只有10 bp不到,所有“外显子”看起来一样大,而且功能分类太多,一个结构图上花花绿绿的各种颜色,无法让人一眼get你想表达什么信息。
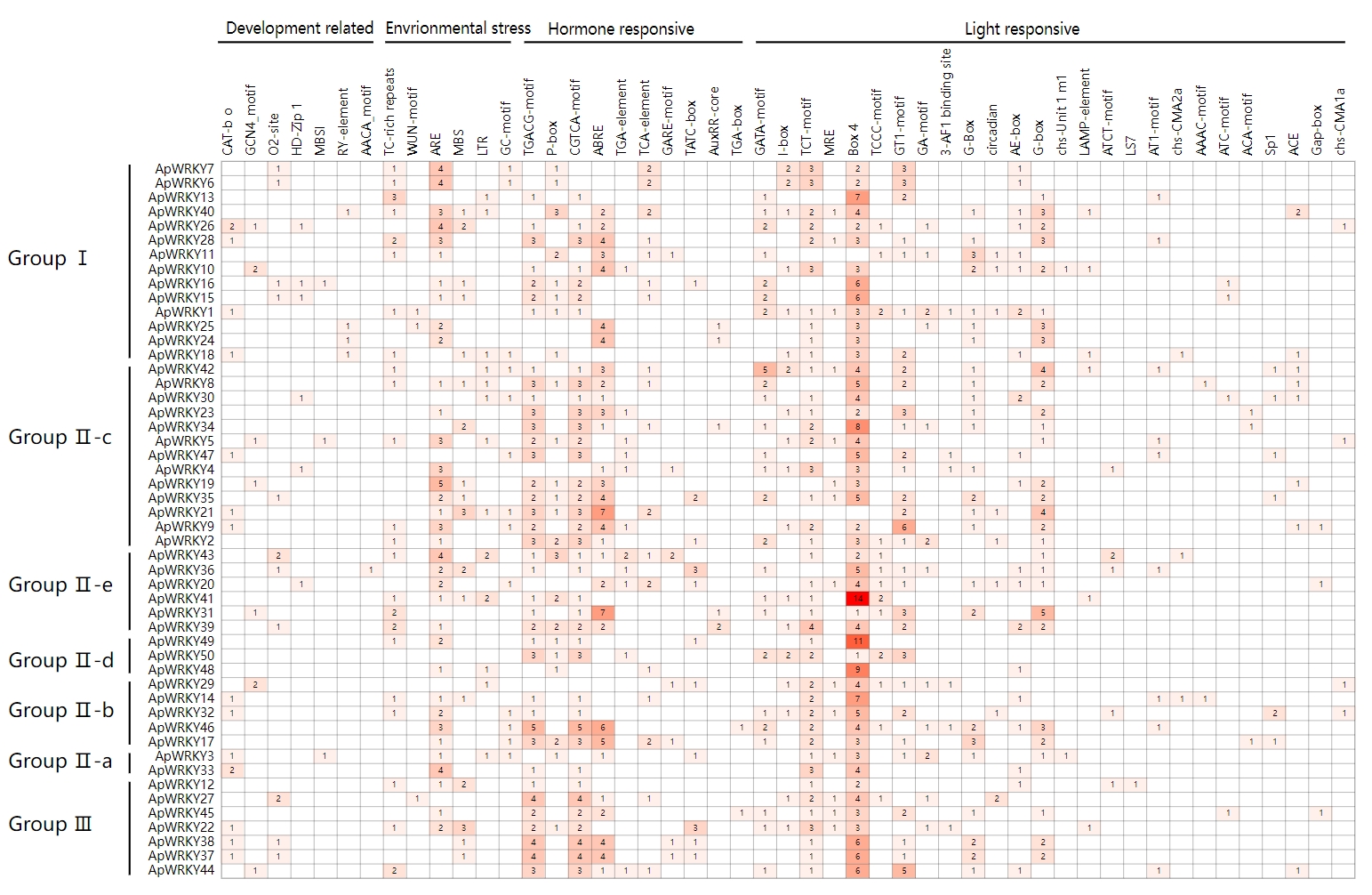
我觉得还不如直接统计预测的各种顺式元件是什么功能,上面的四种功能分区分别有多少成员,这种让人一眼可以得到有效信息的统计图更实在。因此,位置信息就没有任何作用了,对表格进一步精简,只保留A列、B列和H列即可,重命名为:cis.classification.tab.xls

我这里对显示的基因有顺序要求,我需要将同一个亚家族的基因放在一起进行比较。因此,需要一个给基因排序的文件order.txt:
就是简单给基因进行手动排序,方便后续AI添加x轴和y轴的分类。作图使用R,代码如下:
1 | library(tidyverse) |
主图出来后,简单用AI添加x轴和y轴的分类即可(x轴顺式元件名称是按照四个功能类型分组的),如下:
同样,还可以做一个堆积柱状图来统计四种功能的顺式元件个数:
1 | # 重设因子水平,做第二个水平方向的堆积柱状图 |
出图如下:

做这种水平的堆积图只是为了方便拼接上面的统计图(因为Y轴基因顺序完全一致),也可以做成正常的堆积柱状图展示,这里就不作多解释了。